In 2020, an estimated 147,950 patients will be diagnosed with colorectal cancer (CRC) in the USA, with 53,200 deaths expected.1 While many patients with CRC are cured, metastatic CRC (mCRC) is still a prevalent disease, and systemic therapy options have markedly improved outcomes in patients with unresectable metastatic disease in the last several decades. Cytotoxic chemotherapy options have largely been a mainstay of therapy, with fluoropyrimidine, oxaliplatin and irinotecan being the early standards of therapeutic options. However, with the advent of the anti-vascular endothelial growth factor (VEGF) and anti-epidermal growth factor receptor (EGFR) therapies in mCRC, additional, unique signal transduction pathways have been discovered and targeted. As a result, over the past two decades, many new treatment options identified by precision medicine have been investigated in CRC, and our armamentarium of treatment options has increased.
Despite these discoveries and increased knowledge of targeted pathways critical for response to therapies, molecular profiling to predict response to therapy is largely underutilized. Current National Comprehensive Cancer Network (NCCN) guidelines recommend all patients with mCRC be tested for KRAS (Kirsten rat sarcoma virus homolog), NRAS (neuroblastoma rat sarcoma virus homolog), and BRAF (v-Raf murine sarcoma viral oncogene homolog B) mutations, as well as microsatellite instability (MSI) or mismatch repair deficiency (dMMR).2 However, a recent study evaluated 1,497 patients with metastatic colon cancer treated in both academic and community oncology practices between 2013 and 2017, and found guideline-aligned biomarker testing rates for RAS, BRAF, and MSI-high (MSI-H)/dMMR to be extremely low at 41%, 43%, and 51%, respectively.3
As the field of genomics advances, improvements in sequencing techniques have led to better detection of potentially druggable genomic alterations. For instance, tissue-based next-generation sequencing platforms now allow rapid, comprehensive molecular profiling of patient tumors, which can directly affect treatment. Additionally, liquid-based biopsies with detection of cell-free DNA can now not only reliably detect small fragments of genetic material shed by tumors into the blood for assistance in initial therapeutic decision making for mCRC, but cell-free DNA assays can also detect tumor heterogeneity and acquired resistance to therapies.4–6
In this article, validated and emerging genomic targets and their therapeutic correlates in mCRC will be discussed. We focus both on the validated targets and their concordant targeted therapeutic options, as well as emerging targets and areas of investigation within these actionable genomic alterations. We also describe a novel screening platform with targeted-therapy companion trials that will hopefully increase our understanding of the biology behind tumor response and resistance to therapies, as well as improve patient outcomes in refractory mCRC.
Actionable genomic targets in metastatic colorectal cancer
BRAF V600E mutant colorectal cancer
Mutations in BRAF V600E are found in approximately 8–10% of mCRCs, and this finding portends a worse overall prognosis for patients, both in terms of survival as well as responsiveness to cytotoxic chemotherapy.7,8 BRAF is a key downstream effector of RAS in the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signal transduction pathway, and is known to be an oncogenic driver in these tumors.9 Interestingly, not all BRAF mutations seen in CRC confer a worse prognosis; non-V600 BRAF-mutant CRC appears to be a distinct molecular subtype with an improved prognosis.10 For the purpose of this discussion, however, we will focus on BRAF V600E-mutant mCRC.
The BRAF V600E tyrosine kinase inhibitor, vemurafenib, has been explored in the clinical setting on the basis of its antitumor activity seen in preclinical models of BRAF-mutant CRC.11 Patients with BRAF-mutant mCRC were treated in a phase II study of vemurafenib 960 mg twice daily.12 While 1/21 (4.8%) patient had a partial response and 7/21 (33.3%) had stable disease by Response Evaluation Criteria in Solid Tumors (RECIST), median progression-free survival (PFS) was a disappointing 2.1 months. Similar disappointing results were also seen with monotherapy utilizing the BRAF inhibitor encorafenib.13
Given the failure of BRAF inhibitor monotherapy in this population, the combination of vemurafenib, irinotecan and cetuximab was then investigated in patients with refractory BRAF-mutant mCRC.14 The rationale for this combination was driven by the knowledge that BRAF inhibition suppresses ERK-mediated negative feedback on EGFR activity, leading to EGFR activation and ultimately CRAF-mediated reactivation of the MAPK/ERK pathway; EGFR signaling is therefore critical in bypassing BRAF inhibition.15,16 The phase Ib study of vemurafenib, irinotecan and cetuximab demonstrated an improved response rate of 35% (6/17 evaluable patients) with the triplet therapy, and a median PFS of 7.7 months. A follow-up randomized phase II study of irinotecan and cetuximab with or without vemurafenib in this cohort demonstrated an improvement in PFS with vemurafenib (hazard ratio [HR] 0.42, 95% confidence interval [CI] 0.26–0.66; p<0.001) and median PFS of 4.4 months (95% CI 3.6–5.7) versus 2.0 months (95% CI 1.8–2.1).17
Further study of this CRC subtype revealed that combined inhibition of BRAF and MEK (MAPK kinase) was more effective than combined inhibition of BRAF and anti-EGFR therapy.15,18,19 As a result, the phase III BEACON study (ClinicalTrials.gov identifier: NCT02928224) was undertaken, in which 665 patients with pretreated BRAF V600E-mutant mCRC were randomized to the following treatments: the BRAF inhibitor encorafenib, MEK inhibitor binimetinib, and anti-EGFR therapy cetuximab; encorafenib or cetuximab; or investigators’ choice of cetuximab and irinotecan or cetuximab with folinic acid, fluorouracil and irinotecan (FOLFIRI).20 Median overall survival (OS)
was 9.0 months in the triplet-therapy group, 8.4 months in the doublet group, and 5.4 months in the control group, though the study was powered for comparison between the triplet and control groups only (HR 0.52, 95% CI 0.39–0.70; p<0.001). Response rate for the triplet group was 26% compared with 2% in the control group (p<0.001). Of note, grade 3 or higher adverse events occurred in 58% of patients in the triplet group, 50% of the doublet group, and 61% of the control group.
While the BEACON trial results are encouraging for the treatment of patients with BRAF-mutant mCRC, the significant toxicities of these therapies and the dearth of subsequent effective therapies in a poor prognostic patient cohort emphasize the need for additional therapies for these patients. Currently, the single-arm phase II ANCHOR-CRC study of encorafenib, binimetinib and cetuximab in previously untreated patients with BRAF-mutant CRC, is investigating front-line triplet therapy for this subset of patients (ClinicalTrials.gov identifier: NCT03693170; Table 1). Furthermore, and perhaps most importantly, investigation of potential resistance mechanisms to these targeted therapies are underway and hold promise to improve patient outcomes with thoughtful targeting of critical oncogenic pathways.
HER2-amplified/overexpressed colorectal cancer
Another emerging and clinically relevant target in mCRC is that of human epidermal growth factor receptor 2 (HER2) oncogene amplification or overexpression, which is present in approximately 5% of patients with RAS wild type mCRC, distinct from mutations in ERBB2, and defined by immunohistochemical staining and/or in situ hybridization.21 Preclinical studies of HER2 amplification in KRAS/NRAS/BRAF wild-type patient-derived CRC xenografts demonstrated resistance to anti-EGFR therapies,22,23 and poorer patient outcomes were seen with anti-EGFR therapy in the clinical setting as well.24 However, targeting the HER2 oncogene preclinically with the combination of the anti-HER2 therapies trastuzumab and lapatinib showed tumor response.25
On the basis of these preclinical data, the open-label phase II HERACLES study utilized the combination of the anti-HER2 monoclonal antibody trastuzumab and the tyrosine kinase inhibitor lapatinib in patients with refractory KRAS exon 2 wild-type, HER2-positive mCRC (EudraCT number: 2012-002128-33).26 In this study, the HER2 diagnostic criteria were defined as tumors HER2 3+ by immunochemistry, or those HER2 2+/fluorescence in situ hybridization positive.21 Patients were treated with intravenous trastuzumab 4 mg/kg loading dose followed by
2 mg/kg once weekly, and oral lapatinib 1,000 mg daily, with a primary endpoint of objective response. A total of 914 patients with KRAS exon 2 wild-type were screened, and 48 (5%) patients with HER2-positive tumors were identified. Eight of 27 treated patients (30%, 95% CI 14–50), all of which were heavily pre-treated, achieved an objective response, including one complete response and seven partial responses; 12 (44%, 95% CI, 25–63) patients had stable disease on therapy. A total of 6/27 (22%) of patients had grade 3 adverse events in this study. Notably, the majority (88%) of responding patients had tumors that were HER2 3+ by immunohistochemistry. Furthermore, median PFS of patients with tumors harboring a HER2 gene copy number greater than 9.45 was markedly improved at 29 weeks (95% CI 19–43) when compared with those with HER2 copy number <9.45; these patients had median PFS of 16 weeks (95% CI 3–17). All of the responding patients had HER2 gene copy numbers >9.45, suggesting that this biomarker may not be a strictly binary one in terms of its predictive nature for response to anti-HER2 therapy.
Another study of dual anti-HER2 targeted therapy in mCRC, the MyPathway phase IIa trial, investigated the use of anti-HER2 antibodies trastuzumab (8 mg/kg loading dose, then 6 mg/kg every 3 weeks intravenously) and pertuzumab (840 mg loading dose, then 420 mg every 3 weeks intravenously) in patients with HER2-amplified, treatment refractory mCRC (ClinicalTrials.gov Identifier: NCT02091141).27 A total of 57 patients with HER2+ tumors by fluorescence in situ hybridization, chromogenic in situ hybridization, copy number gain by next-generation sequencing or immunohistochemistry (3+) were treated, with an overall objective response rate of 32%; one patient (2%) had a complete response and 17 (30%) had partial responses. Estimated median PFS was 2.9 months (95% CI 1.4–5.3) and median OS was 11.5 months (95% CI 7.7–not estimable). Median duration of response was 5.9 months (95% CI 2.8–11.1). Interestingly in this study, co-occurring KRAS mutations (accounting for 23% of treated patients) were associated with lower response to dual anti-HER2 therapy, as were mutations in PIK3CA.
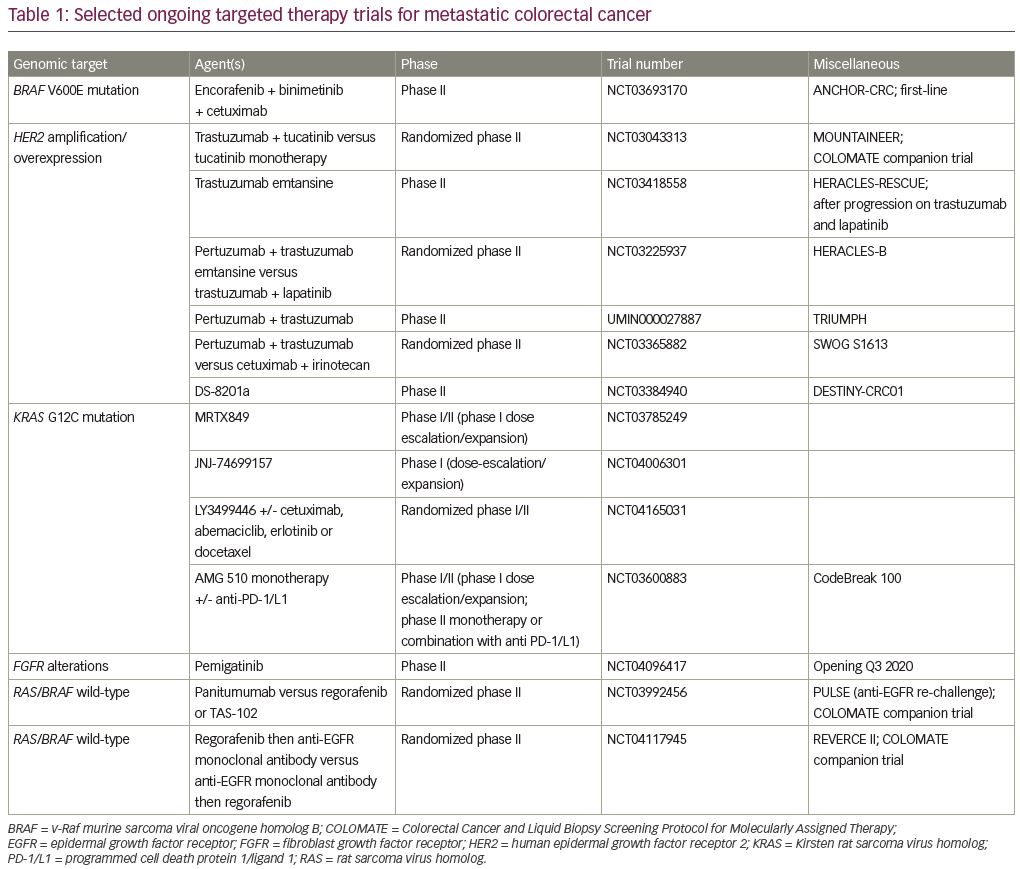
Other ongoing HER2-directed therapeutic studies in mCRC are currently of significant interest, including MOUNTAINEER (trastuzumab and tucatinib; ClinicalTrials.gov identifier: NCT03043313),28 HERACLES-RESCUE (trastuzumab emtansine after progression on trastuzumab and lapatinib; NCT03418558), HERACLES-B (pertuzumab plus trastuzumab emtansine; NCT03225937); TRIUMPH (pertuzumab plus trastuzumab; UMIN000027887),29 SWOG S1613 (pertuzumab and trastuzumab versus cetuximab and irinotecan; NCT03365882), and a study of DS-8201a (NCT03384940), among others (Table 1).
The phase II study of trastuzumab and the tyrosine kinase inhibitor tucatinib, entitled MOUNTAINEER, is particularly promising, with recent results presented at the European Society for Medical Oncology 2019 Annual Congress in Barcelona, Spain.28 In this study, the objective response rate to therapy was an impressive 52.2% (95% CI 30.6–73.1), with median duration of response of 10.4 months (95% CI 6.0–not estimable). Median PFS was 8.1 months (95% CI 3.8–not estimable) and median OS was 18.7 months (95% CI 12.3–not estimable) in a heavily pre-treated mCRC population. These ongoing studies will hopefully help elucidate the optimal HER2-targeted approach and sequencing of therapy, as HER2 has clearly become a validated therapeutic target in mCRC.
NTRK fusions in colorectal cancer
Though uncommon, fusions involving one of three tropomyosin receptor kinase (TRK) proteins encoded by the neurotrophic receptor tyrosine kinase genes NTRK1, NTRK2 and NTRK3, occur in CRC, and this is now an actionable genomic finding in many tumor types. NTRK fusions can be identified in a tumor by DNA- or RNA-based next-generation sequencing, pan-TRK immunohistochemistry, reverse transcription polymerase chain reaction (PCR) or fluorescence in situ hybridization.30,31 In a study of the highly-selective small molecular inhibitor of all three TRK proteins, larotrectinib, in patients with NTRK fusions, 4/55 treated patients (7%) had CRC.32 The overall response rate in the multi-tumor type population was an impressive 80% (95% CI 67–90), but only 2/4 (50%) of the patients with CRC had an objective response. At the time of publication, median duration of response and PFS for the cohort as a whole had not been reached. As a result of these data, larotrectinib was granted United States Food and Drug Administration (FDA) approval in November 2018 for the treatment of patients with solid tumors harboring an NTRK gene fusion without a known acquired resistance mutation.
Subsequently, the TRKA/B/C, ROS1 and anaplastic lymphoma kinase inhibitor, entrectinib, was studied in three trials (ALKA-273-001, STARTRK-1, and STARTRK-2), with a total of 54 adult patients treated and evaluable for efficacy (ClinicalTrials.gov identifiers: NCT02097810 and NCT02568267, and EudraCT number: 2012–000148–88).33 A total of 31 patients (57%, 95% CI 43.2–70.8) had an objective response, including four (7%) achieving a complete response. In this patient cohort, four (7%) patients had CRC, and two of these patients had an objective response to therapy.
While tumor responses to TRK inhibitors seem very promising, it must be remembered that this genomic alteration is extremely uncommon in CRC. A recent cancer-wide screening program of 26,000 patients showed an overall prevalence of TRK fusions to be 0.28% (74/26,312).34 Of the TRK–fusion positive patients in this cohort, eight (10.5%) had CRC, but the prevalence of TRK fusions overall in CRC was 0.35 (95% CI 0.15–0.68), or 8/2,306. Importantly, while other oncogenic drivers were generally seen less frequently with TRK fusions, an exception to this was concurrent MSI in CRC. A study by Cocco et al. suggests that CRCs with hypermethylated MLH1 promoter and wild-type BRAF/KRAS are enriched for targetable kinase fusions such as TRK.35
Next-generation TRK inhibitors are currently under investigation, and improvement in clinical responses and overcoming of acquired resistance have now been observed. Due to its low prevalence as a target and apparent decrease in tumor responsiveness to therapy in CRC, however, TRK fusions are unlikely to be a dominant therapeutic target in this tumor type.
Microsatellite instability-high/deficient mismatch repair colorectal cancer
Another critical genomic alteration found in a minority (3–4%) of mCRCs is that of MSI, or dMMR. Identified by immunohistochemistry analysis revealing the loss of mismatch repair gene proteins MLH1, PMS2, MSH2 and MSH6 (dMMR), PCR of Bethesda markers, or next-generation sequencing, dMMR/MSI-H is the strongest predictive biomarker of response to immunotherapy in CRC.36,37 This is in contrast to programmed death ligand 1 (PD-L1) expression and tumor mutational burden, among other potential biomarkers, which have not been clinically validated as predictive in mCRC. A study led by Le et al. investigated the effects of programmed cell death protein 1 (PD-1) blockade with pembrolizumab in dMMR tumors independent of the tissue of origin, but including CRC as a dominant type.38 In this phase II study, a total of 86 patients with dMMR were treated with pembrolizumab 10 mg/kg intravenously every 2 weeks, with objective radiographic responses seen in 53% (46/86, 95% CI 66–85) and 21% (18/86) achieving a complete radiographic response. Of note, average time to any response was 21 weeks, but by the time of publication, neither median PFS nor OS had been reached. Patients with CRC had an objective response rate of 52% (95% CI 36–68), similar to patients without CRC (54%, 95% CI 39–69).
Another phase II study, CheckMate 142, confirmed similar findings in patients with dMMR/MSI-H mCRC treated with the PD-1 immune checkpoint inhibitor nivolumab (ClinicalTrials.gov identifier: NCT02060188).39 In this trial, patients were given nivolumab 3 mg/kg intravenously every 2 weeks, with a primary endpoint of objective response. A total of 23 of 74 patients (31.1%, 95% CI 20.8–42.9) achieved an objective response, with median duration of response not reached at the time of publication. Importantly, eight patients had responses lasting 12 months or longer (event rate 86%, 95% CI 62–95).
Though these response rates are impressive in heavily pretreated CRC patients, there are still patients with MSI-H/dMMR mCRC who do not respond to PD-1 blockade monotherapy. In CheckMate-142, another cohort of patients were treated with nivolumab plus ipilimumab, an antibody targeting the cytotoxic T-cell lymphocyte antigen-4 (CTLA-4) checkpoint receptor.40 With the combination regimen, 55% of 119 patients with mCRC achieved an objective response, with disease control rate for at least 12 weeks of 80%, though toxicities were greater than with PD-1 monotherapy. With the nivolumab/ipilimumab combination, PFS rate at 12 months was 71%, and OS rate at 12 months was 85%.
As these clinical data show, PD-1 ± CTLA-4 blockade benefits many patients with MSI-H/dMMR mCRC, but further studies are needed to understand and overcome mechanisms of innate and acquired resistance to immunotherapy, and to optimally sequence these agents. Additionally, since MSI/dMMR is seen in a small minority of patients with mCRC, and immune checkpoint inhibition does not benefit patients with microsatellite stable disease, more treatment options are needed for patients with mCRC not eligible or not responsive to this therapeutic class of agents.
Genomic targets under investigation in metastatic colorectal cancer
KRAS G12C mutations in colorectal cancer
As comprehensive molecular platforms become more commonly used in oncologic practice, additional druggable targets are being identified in CRC. Furthermore, well known targets previously thought to be “undruggable” are now experiencing a resurgence in interest as newer targeted therapies are developed. One example of this phenomenon is that of KRAS inhibition. Mutations in the RAS oncogene are the most common activating mutation in human cancer, with KRAS mutations being much more prevalent than HRAS and NRAS.41–43 While a very prevalent gene family with mutations commonly found in cancers such as CRC, KRAS has been a difficult drug target due to its structure, which prevents efforts to develop nucleotide-competitive inhibitors and identification of high-affinity allosteric inhibitors.41,44 In 2013, Ostrem et al. developed a covalent inhibitor to target the reactive cysteine-12 of KRAS G12C.45 Subsequent efforts by Amgen to develop a targeted covalent inhibitor of KRAS G12C suitable for clinical application resulted in the discovery of the AMG 510 compound, a highly potent and selective covalent inhibitor of KRAS G12C that has subsequently been tested in the clinic.44 Additional efforts by others to develop KRAS G12C inhibitors are also underway (Table 1).
A phase I dose-escalation trial of AMG 510 in patients with locally advanced or metastatic KRAS G12C mutant solid tumors is currently underway, with the compound showing preliminary clinical efficacy in non-small cell lung cancer (5/10 patients with partial responses by RECIST 1.1) and CRC (14/19 patients with stable disease as best response).46 While development of a KRAS inhibitor is an exciting advance in this field, patients with KRAS G12C-mutant CRC are relatively uncommon, with only approximately 3% of patients harboring this mutation.46 Furthermore, the lack of objective responses seen in the CRC cohort compared with other tumor subtypes is somewhat disappointing. However, this is an active and early phase of investigation, and further preclinical data suggest potential efficacy in combination with inhibition of other targets such as MEK and PD-1.47
COLOMATE—an avenue for investigating promising genomic targets in metastatic colorectal cancer
As additional genomic targets are identified for potential therapeutic use in mCRC, robust clinical trials must be performed to validate these targets. The targets currently under investigation (and many of which have already been validated) are of low prevalence in mCRC. Therefore, single-institution clinical trials with requirements of particular genomic alterations are becoming less feasible. Furthermore, even multi-institution genotype-directed trials can be unwieldy and inefficient. To overcome these obstacles, efforts are underway to most efficiently test new therapeutic, genotype-directed treatments. One such example is that of a platform entitled Colorectal Cancer and Liquid Biopsy Screening Protocol for Molecularly Assigned Therapy, or COLOMATE (Figure 1).48 This phase II umbrella screening protocol is sponsored by the Academic and Community Cancer Research United (ACCRU) consortium. It utilizes a blood-based targeted next-generation sequencing panel (Guardant360®, Guardant Health, Redwood City, CA, USA) to identify actionable genomic alterations in patients with mCRC and to assess the impact of molecularly assigned therapy accordingly under its individual companion studies. Companion studies are dynamic and under active development according to emerging scientific discoveries and pharmacologic drug development. The primary objectives of this study are to perform blood-based genomic profiling on patients with treatment-refractory mCRC to facilitate accrual to molecularly assigned therapies, and to obtain patient-matched tumor tissue and cell-free DNA from peripheral blood to facilitate clinically annotated genomic analyses. Secondary objectives are to explore mechanisms of acquired resistance to molecularly assigned therapy and to explore the correlation between cell-free DNA mutational burden and clinical outcomes. The anti-HER2 therapy mCRC trial MOUNTAINEER, as described above, is one example of a companion trial included within COLOMATE; other selected companion trials are highlighted below and in Figure 1.
Fibroblast growth factor receptor alterations in colorectal cancer
The fibroblast growth factor receptor (FGFR) family of receptors (FGFR1–4) and respective 18 ligands are an important signal transduction pathway in cancer, with FGFR pathway activation promoting cellular proliferation, survival and migration, as well as growth arrest.49,50 Dysregulation of the FGFR pathway can lead to the establishment and progression of cancer, and genetic alterations in the FGFRs have been described in many tumor types.51,52 These alterations include activating mutations, translocations, and gene amplification resulting in ligand-independent, constitutive receptor activation or aberrant ligand-dependent signaling through FGFRs.
FGFR inhibition has been studied in tumor types other than CRC, including urothelial carcinoma and cholangiocarcinoma, among others. In fact, FDA approval was granted in 2019 to erdafitinib, an oral FGFR1–4 inhibitor, in the treatment of locally advanced or metastatic urothelial carcinomas harboring FGFR2 or FGFR3 mutations.53,54 Multiple FGFR inhibitors such as pemigatinib, derazantinib and TAS-120 have been investigated in biliary tract cancers with intriguing results.55–57
Molecular alterations in FGFR have been described in CRC in multiple studies, with an overall prevalence of approximately 3–8%.58–60 Given the relative prevalence of mCRC, this could be a significant target. Therefore, a multicenter, phase II, single-arm trial within the COLOMATE umbrella screening platform has been designed utilizing the oral FGFR1–3 inhibitor, pemigatinib, in patients with mCRC harboring active FGFR alterations, and enrollment will begin nationally in Q3 2020 (ClinicalTrials.gov identifier: NCT04096417; Table 1). Initially, this trial will enroll patients with FGFR1–3 alterations of all types (translocations, point mutations, and amplifications) to investigate the role of FGFR inhibitors in this subset of mCRC.
Epidermal growth factor receptor re-challenge in colorectal cancer
In RAS wild-type mCRC, anti-EGFR therapy is often an important therapeutic option; however, sensitive tumors eventually develop resistance to these agents through a variety of mechanisms, including the acquisition of RAS mutations.61–64 After anti-EGFR withdrawal, these clones can exponentially decay,65 and this can be detected non-invasively with circulating tumor DNA testing.66,67 Retreatment of patients with mCRC with anti-EGFR therapies can have benefit,68,69 but the ideal timing, benefit and sequencing of retreatment is not known.
To further investigate this concept, the phase II COLOMATE companion study entitled PULSE is a randomized, open-label study of panitumumab re-challenge versus standard therapy after progression on anti-EGFR therapy in patients with metastatic BRAF/RAS wild-type CRC (ClinicalTrials.gov identifier: NCT03992456; Table 1). Eligible patients must have had treatment with a prior anti-EGFR antibody for at least 4 months and cannot have detectable gene BRAF, EGFR, HER2, KRAS, MET, or NRAS mutations (mutant allele frequency >0.5%) or amplifications at enrollment. Importantly, cell-free DNA will be studied throughout the trial to monitor for dynamic genomic changes in response to therapy. This study is poised to delineate optimal sequencing of anti-EGFR therapy in mCRC beyond initial acquired resistance.
Anti-vascular endothelial growth factor and anti-epidermal growth factor receptor sequencing in colorectal cancer
In addition to timing of anti-EGFR therapy in RAS/RAF wild type mCRC, the initial sequencing of cetuximab or panitumumab in these patients is unknown. The randomized phase II REVERCE trial investigated the efficacy and safety of the anti-VEGF tyrosine kinase inhibitor regorafenib followed by cetuximab with or without irinotecan (R–C arm), compared with cetuximab with or without irinotecan followed by regorafenib (C–R arm), in patients with RAS wild-type mCRC whose tumors had progressed on fluoropyrimidine, oxaliplatin and irinotecan (Table 1).70 A total of 101 patients were randomized and eligible for efficacy analysis, with the primary endpoint of OS. Median OS for R–C was markedly better than the standard C–R (OS 17.4 versus 11.6 months, p=0.0293; HR 0.61, 95% CI 0.39–0.96). Additionally, emerging alterations in RAS, BRAF, EGFR, HER2 and MET were more commonly seen after exposure to anti-EGFR therapy than regorafenib. Despite these intriguing data, the study results were considered only hypothesis-generating as the study ended early for funding reasons and was not fully accrued.
Given these surprising results, the sequencing of anti-VEGF and anti-EGFR therapies in patients with mCRC should be further explored. REVERCE II, a COLOMATE companion trial, is a randomized phase II study of regorafenib followed by anti-EGFR monoclonal antibody therapy versus the reverse sequencing for patients with mCRC previously treated with fluoropyrimidine, oxaliplatin and irinotecan (ClinicalTrials.gov identifier: NCT04117945; Table 1). Given the unique characteristics of COLOMATE, which encourage dynamic, ongoing cell-free DNA analysis throughout the patient’s treatment course, this study should help elucidate the biologic underpinnings of anti-VEGF/anti-EGFR therapeutic responses depending on treatment sequencing.
Other COLOMATE companion studies in development
In addition to the studies described above, multiple other companion studies are under development through COLOMATE, including trials targeting patients with mCRC whose tumors harbor MET amplification, PIK3CA mutations, EGFR amplification and others (Figure 1). Furthermore, COLOMATE incorporates a “no actionable change” arm, which allows enrollment and treatment of patients whose tumors do not contain any currently actionable genomic alterations; this allows for inclusion of robust and scientifically meritorious non-targeted agents and combinations in the companion trials, and maximizes treatment of patients enrolled on the COLOMATE screening platform. This approach is designed to dynamically respond to emerging scientific discoveries and efficiently test new hypotheses for improving patient outcomes with mCRC.
Conclusions
In conclusion, molecular profiling is a critical component in the identification of optimal treatment selection and sequencing in mCRC. Currently validated targets include KRAS, NRAS, BRAF, HER2, MSI/dMMR, and NTRK, and targeted therapy selection according to the status of these genomic alterations is defined. Numerous other targets, some of which have been described here, are under investigation as potential predictive biomarkers of response to targeted therapies. As additional targets and treatment options are discovered and validated, an understanding of the biological underpinnings of sensitivity and primary or acquired resistance to these targeted agents will be needed. Novel molecular profiling mechanisms and clinical trial platforms, particularly when mCRC patients’ tumors are profiled early in their treatment course, hold promise to most efficiently and accurately identify key oncogenic drivers and appropriate treatments in mCRC.