Liquid biopsy: Initial concepts and application in solid tumours
The US National Cancer Institute defines liquid biopsy (LB) as ‘a test done on a sample of blood to look for cancer cells from a tumour that are circulating in the blood or for pieces of DNA from tumour cells that are in the blood’.1 In recent decades, the growth of precision medicine and the advancement of genomic and molecular profiling techniques have positioned LB as a highly sensitive, versatile, reproducible and cost-effective method for real-time biomarker evaluation.2 LB also bypasses pre-analytical variables, minimizes sample depletion and reduces pitfalls related to tumour heterogeneity, a major limitation of tissue biopsies, which are currently considered the gold standard in diagnosis.3
Circulating tumour cells (CTCs) were first reported in 1869 by Thomas Ashworth.4 Following this, improvements in technology contributed to the identification of circulating tumour DNA (ctDNA), circulating cell-free DNA (cfDNA), exosomes and microRNA (miRNA).5 CTCs are tumour-originated cells that have been shed into the bloodstream. Their presence is directly associated with treatment outcomes and overall survival (OS), and their clinical utility resides in the ability to provide insight into tumour biology through molecular characterization. CfDNA refers to fragments of DNA that are shed into the bloodstream following apoptosis of haematopoietic cells, while ctDNA consists of fragments of DNA shed exclusively by cancer cells. The detection of circulating tumour RNA and miRNA enables the detection of tumour-specific gene expression profiles. Exosomes are microvesicles released from tumour cells due to the fusion of multivesicular bodies with the plasma membranes. Their use as an oncological biomarker has grown with the study of molecular information contained in their proteins and nucleic acids (DNA, mRNAs and miRNAs). In addition to that, miRNA plays a role in disease progression as it is known to prompt angiogenesis and the further development of metastatic disease.6
Currently, LB is being developed for a range of clinical applications, including screening and early diagnosis, triage, staging, risk stratification, follow-up and guiding systemic treatment decisions, as demonstrated in Figure 1. Additionally, ongoing discussions focus on cancer-personalized profiling via deep sequencing, its role as a biomarker for immunotherapy response, exemplified by the PROphet platform (a proteomics-based test used for risk stratification and decision-making in patients with non-small cell lung cancer) and the development of pan-tumour diagnostic tests.7–9
Figure 1: Liquid biopsy applications

Schematic representation of liquid biopsy applications throughout the natural disease course of melanoma, illustrating quantitative tumour burden tracking via serial testing. The number of vials is proportionally scaled to represent the corresponding levels of circulating tumour cells at each disease stage.
Although tissue sample biopsy remains the gold standard, the use of LB has become increasingly attractive due to several advantages over tissue sampling, including the fact that it is a less invasive andcost-effective procedure, with virtually no clinical risk of complications other than the one inherent to venipuncture. Additionally, LB can capture or even overcome the phenomenon of tumour heterogeneity, providing information with potential therapeutic implications. The method of sample storage also favours the use of LB, as the substances used for the preservation of tissue samples can interfere with the quality and result of molecular analyses, which is often the reason for performing the biopsy.10,11
In solid tumours, only a few LB-based methods have been validated and incorporated into clinical practice. However, unsolved challenges result from the complexity of personalizing biomarkers for solid tumours, as they are continuously affected by genetic and environmental factors, which complicates clinical interpretation. Well-established examples of LB use in solid tumours include lung and colorectal cancers, where ctDNA-based diagnostic markers have already been approved by the US Food and Drug Administration, and the CELLSEARCH® CTC test (Menarini Silicon Biosystems, a subsidiary of the Menarini Group, Florence, Italy), which is used as a diagnostic tool for metastatic colon, breast and prostate cancers.12
Venues for exploring liquid biopsy in melanoma
Melanoma survival trends present a paradox: while prognosis generally worsens with increased tumour thickness, thin melanomas (T1/T2) account for over half of long-term melanoma-specific deaths.13,14 Moreover, melanoma incidence is rising globally, especially among thin tumours, likely due to enhanced public education, more proactive screening and prevention efforts. It is critical to not only view thin melanomas as a public health issue but also to recognize the limitations in their stratification and prognostic classification, particularly when relying on traditional methods such as tissue biopsy.15 Tumour heterogeneity and plasticity, driven by the microenvironment, are key factors to this discrepancy. LB may be a crucial tool in addressing this issue, enabling more precise identification of patients with thin melanomas at high biological risk, thus facilitating tailored surveillance and early intervention strategies.
Stage II and stage III melanoma continues to draw significant attention, given the unresolved dilemmas. Notably, survival outcomes diverge markedly among stage II patients, with 10-year survival rates ranging from 94% in stage IIA to 75% in stage IIC; a prognosis worse than that of stage IIIA (88%) and comparable with stage IIIB (77%). Similarly, stage III cohorts exhibit pronounced variability as stage IIID outcomes mirror those of advanced metastatic disease.16 Concerningly, this diversity is still not adequately managed in clinical practice, with similar treatment and surveillance protocols being applied across these groups.17 This poses a significant risk of overtreating patients with localized or locally advanced disease, exposing them to severe and sometimes irreversible immunotherapy toxicities, when they could potentially be cured with surgery alone.
Melanoma cells exhibit high plasticity, rapidly adapting to changes in the microenvironment and treatment pressures.18 A significant challenge lies in the uncertainty around the optimal treatment duration for advanced melanoma, with limited evidence on when to discontinue therapy in those with complete response. Additionally, concerns about the effectiveness of current surveillance protocols persist. While over half of the first recurrences become systemic in stage III, nearly 50% of melanoma recurrences are detected through patient, family or physician reports, surpassing 32% of recurrences identified by imaging methods.19 LB offers an opportunity to shift from reactive follow-up to proactive precision monitoring based on the molecular risk of each patient. This issue is even more critical in cases such as amelanotic and uveal melanoma (UM), which require more innovative care strategies.
Screening, early diagnosis and triage
Although most melanoma cases are detected at an early stage, up to 15% of patients will be diagnosed with either nodal involvement or metastatic disease.20 Given the considerable inferior survival outcomes in these scenarios, the use of LB, particularly in high-risk populations, can identify subclinical disease and allow for early interventions, impacting morbidity, mortality, metastasis development risk and costs. In their study, De Giorgi et al. applied a filtration technique to detect CTCs – cancer cells that have detached from a primary or metastatic tumour and circulate in the bloodstream – directly in 140 subjects, including 87 patients with cutaneous melanoma, 10 undergoing surgery for melanocytic nevi, 5 with non-melanoma skin tumours and 38 healthy volunteers.21 In this series, CTCs were detected and immunohistochemically verified in 29% of patients with primary invasive melanoma, with none of the controls showing detectable melanoma CTCs. This study was pioneering in demonstrating the potential of CTCs as diagnostic biomarkers for melanoma, even in high-risk patients with other cutaneous lesions in the differential diagnosis.
Van Laar et al. studied the circlating miRNA signature MEL38 in 372 patients with invasive melanoma and 210 controls, which included primary or metastatic melanoma, melanoma in situ, nonmelanoma skin cancer and benign nevi.22 Using a MEL38 cutoff of >5.5, 95% of patients were correctly diagnosed, with 93% sensitivity and 98% specificity. In conclusion, the circulating MEL38 signature may assist in diagnosing invasive melanoma versus other conditions with lower or negligible mortality risk, which are common in clinical practice and often lead to unnecessary invasive procedures and diagnostic-related anxiety.
Although useful, LB is not yet a perfect diagnostic method. ctDNA levels correlate with tumour stage and volume, but patients in the early stage or those in screening protocols may have ctDNA levels below detection thresholds.23 For example, one study found a low detection rate of BRAFV600E mutation through circulating free DNA analysis in patients with early-stage melanoma.24 Furthermore, classic driver mutations can also appear in non-malignant cells, especially in older individuals. For instance, clonal haematopoiesis with somatic mutations was found in 10% of individuals over 65 years, but with a low risk of progression to haematologic cancer (1.0% per year).25 Some melanoma characteristic mutations can also be found in benign conditions, such as BRAF in nevi or FGFR3 in seborrheic keratosis, and may act as a bias in-making.26 Moreover, detecting cancer-associated mutations in cfDNA at a preclinical stage does not necessarily confirm the presence of cancer, highlighting the need for more careful patient selection. It appears to be a valuable tool in high-risk clinical scenarios or when there is diagnostic uncertainty regarding benign skin lesions, as well as for long-term monitoring of patients with numerous or atypical nevi, where dermoscopy and digital mapping may fall short.27
Staging, risk stratification and follow-up
Staging and risk stratification in melanoma are traditionally based on clinical, histopathological and laboratory criteria in accordance with the American Joint Committee on Cancer (AJCC) guidelines.28 In general, elements such as depth, ulceration, lactate dehydrogenase (LDH) levels, regional non-nodal disease, lymph nodes and distant metastasis establish the tumour–node–metastasis classification. However, key characteristics, such as mitotic count and the extent of disease in lymph nodes, significantly affect patient prognosis but are not incorporated into the current classification.29 Adaptations made to the eighth edition of the AJCC staging system included the creation of stage IIID and the reclassification of a subset of stage IIIA patients into IIIB and IIIC.30 While these changes have improved homogeneity within the groups, considerable variability persists. Clinically, this is reflected in a melanoma-specific survival (MSS) rate that ranges from 88% at 10 years (stage IIIA) to only 24% at 10 years (stage IIID) within stage III. Furthermore, it is well established that the first recurrence of melanoma in stage III has a considerable likelihood of being systemic, affecting approximately 61% of cases in stage IIIC patients, significantly worsening their prognosis.19 As a result, there is an unmet need for the development of new risk stratification methods and classification systems in melanoma. In this context, LB has been explored as a complementary approach to the standard methods and has already shown promising results.
Van Laar et al. developed a prognostic risk score algorithm from a novel miRNA signature, MEL12. This genomic signature model is capable of stratifying patients into three distinct groups (low, standard and high risk), which are correlated with MSS. The MEL12 score was also found to have a positive association with the clinical stage and sentinel lymph node biopsy. Moreover, there was no significant association between the risk score and the histological type, which can be an advantage as it shows the method’s ability to avoid potentially confounding covariates when estimating prognosis.22
These findings are consistent with the findings from previous studies. Lee et al. analysed preoperative ctDNA in 119 patients with stage III melanoma and somatic tumour-associated mutation (BRAF, NRAS or KIT). The detection of ctDNA before completing surgical resection was associated with higher clinical stage and LDH levels. Notably, patients whose LB was positive had shorter medium MSS (17.6 versus 49.1 m) and shorter distant metastasis recurrence-free survival (6.2 versus 13.9 m) compared with those with no ctDNA detection.31
In a different study, Váraljai et al. investigated the prognostic and predictive potential of cfDNA concentration in patients with stage III and IV melanoma and wild-type tumours. It is notable that up to 20% of melanoma cases are wild-type, making them unsuitable for ctDNA analyses. In this population, elevated baseline cfDNA levels were linked to an increased risk of tumour progression and mortality, suggesting that high cfDNA concentrations may serve as a marker for identifying patients with poor outcomes, regardless of tumour mutational status. However, this finding should be interpreted with caution as healthy individuals also release cfDNA into circulation.32
Treatment implications
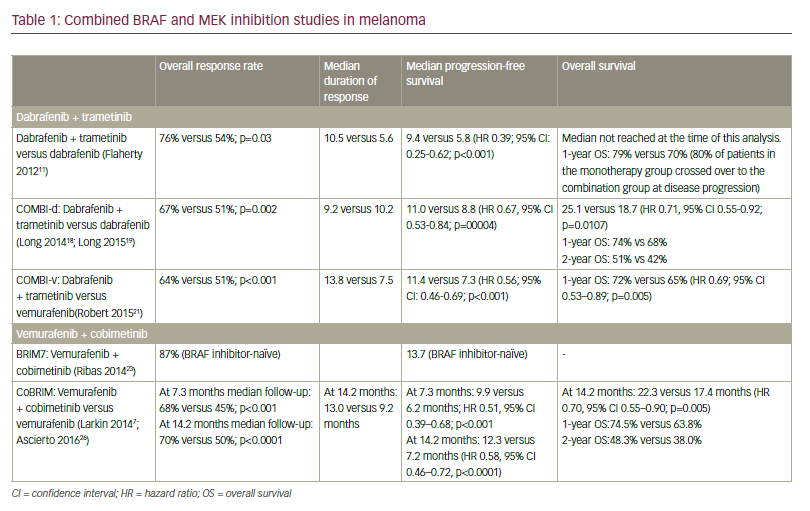
Over the past 15 years, advances in molecular targeted therapy and immune checkpoint inhibitors (ICIs) have significantly improved the treatment and prognosis of advanced melanoma. Previously, cytotoxic chemotherapy achieved only modest efficacy, with response rates of 20–30% and a median OS of 6–8 months. In contrast, combination immunotherapy has led to durable and substantial benefits, as demonstrated in the 10-year follow-up of the CheckMate-067 (Final, 10-Year Outcomes with Nivolumab Plus Ipilimumab in Advanced Melanoma; ClinicalTrials.gov identifier: NCT01844505) trial, which reported a median OS of 71.9 months and objective response rates of 45–58%, setting a new benchmark in melanoma treatment.33
Despite these advancements, critical challenges remain. One major issue is the discrepancy between pathological and radiological responses, particularly in the context of immunotherapy and targeted therapies. Immune-related pathological responses have been observed in biopsy specimens even when imaging suggests stable disease.34 Additionally, pseudoprogression can mimic true disease progression, and metastatic lesions within the same patient often respond heterogeneously, making radiological assessments unreliable indicators of therapeutic efficacy at the cellular or histological level.35 These challenges underscore the need for more precise biomarkers to guide treatment decisions and disease monitoring.
Similarly, cfDNA screening has demonstrated both prognostic and therapeutic value in identifying candidates for molecularly targeted therapies. Santiago-Walker et al. reported that BRAF mutations were detectable in cfDNA from 76% of patients with BRAF V600E and 81% with V600K tumours. Notably, patients with BRAF-mutant tumours but undetectable cfDNA mutations at baseline had longer progression-free and OS than those with detectable mutations, highlighting the potential of cfDNA as both a predictive and prognostic biomarker.36
Beyond the detection of actionable mutations, LB can also assist in evaluating genetic signatures, such as tumour mutational burden (TMB).37 Drusbosky et al. analysed 5,610 solid tumour samples and demonstrated the feasibility of this approach, with blood-based TMB scores exceeding previously reported tissue-based TMB, suggesting that ctDNA may more effectively capture tumour heterogeneity. This further supports its integration into precision oncology, enhancing both therapeutic decision-making and longitudinal disease monitoring.38
Minimal residual disease (MRD) refers to the subset of viable cancer cells that persist following treatment and can ultimately drive disease relapses.39 The detection of MRD through ctDNA analysis has emerged as a promising approach, enabling earlier identification of recurrence compared with conventional surveillance methods, such as periodic imaging and serum LDH assessment. Evidence from various studies indicates that ctDNA-based relapse detection can precede radiological findings by an average of 75 days.40
The sensibility of MRD detection by ctDNA can be increased with the use of longitudinal assessment. Tan et al. demonstrated that the emergence of ctDNA positivity in subsequent measurements following an initial negative result strongly correlates with disease recurrence in the postoperative setting.41 Additionally, ctDNA has shown promise in monitoring response to immunotherapy in both locally advanced and metastatic melanoma.41,42 In a cohort of 18 patients receiving adjuvant ICI, no recurrence was observed in 14 patients with negative LBs. Among the remaining four patients, two exhibited ctDNA clearance after therapy initiation, further supporting its potential as a dynamic biomarker for treatment response.41
Lee et al. investigated the relationship between ctDNA and clinical outcomes in patients with stage IV melanoma receiving monotherapy with programmed cell death receptor 1 (PD-1) inhibitors or in combination with ipilimumab. Baseline and on-treatment samples were collected. Following ctDNA measurement, patients were divided into three distinct groups: group A (undetectable ctDNA at baseline and during therapy), group B (detectable ctDNA at baseline with early conversion to undetectable levels during therapy) and group C (persistently detectable ctDNA from baseline up to 12 weeks after therapy initiation). Patients in groups A and B had longer progression-free survival (PFS) compared with those in group C, suggesting that a negative LB or early conversion to undetectable ctDNA could serve as an independent predictor of treatment response. Other studies have replicated this finding.41,43 The relevance of this discovery lies in the potential to identify patients with unfavourable ctDNA profiles who may benefit from more aggressive treatment strategies.
The utility of ctDNA extends to distinguishing pseudoprogression – which occurs in approximately 20–30% of patients undergoing immunotherapy – from true disease progression. In a cohort of patients treated with PD-1 inhibitors, a 10-fold reduction in ctDNA copy number demonstrated high accuracy in identifying cases of pseudoprogression.44 Furthermore, in patients with stage IV melanoma receiving targeted therapy, elevated baseline ctDNA levels were independently associated with shorter OS, even after adjusting for potential confounding variables. Additionally, baseline ctDNA levels showed an inverse correlation with the duration of response to BRAF/MEK inhibitors. Consistent with findings in patients treated with ICI, conversion of ctDNA status from positive to negative was linked to prolonged PFS and OS.44 This information is summarized in Table 1.41–44
Table 1: Comparative analysis of clinical outcomes by circulating tumour DNA dynamics in melanoma41–44
| Study | Patients | Endpoints and follow-up | Intervention | Outcomes |
| Tan et al.41 | 18 patients with stage III resected melanoma (Melanoma Research Victoria subgroup) | RFS and DMFS; 7 months | Anti-PD1 | RFS of 100% in 14 patients who were ctDNA negative at baseline and in 2 patients who became ctDNA negative postoperatively |
| Lee et al.42 | 76 patients with stage IV melanoma, divided into three groups: A (undetectable ctDNA at baseline), B (elevated ctDNA at baseline but undetectable after therapy) and C (elevated ctDNA at baseline and during treatment) | mOS, mPFS and ORR; 17.5 months | Anti-PD1 or anti-PD1 + ipilimumabe | mPFS not reached in A and B versus 2.7 months in C; mOS not reached in A and B versus 9.2 months in C; ORR of 72% in A, 77% in B and 6% in C |
| Lee et al.44 | 125 patients with stage IV melanoma. Patients were divided into favourable ctDNA profile (undetectable ctDNA at baseline or detectable ctDNA at baseline followed by >10-fold decrease) and unfavourable ctDNA profile (detectable ctDNA at baseline that remained stable or increased) | Early differentiation of pseudoprogression from true progression; 21 months | Anti-PD-1 or anti-PD-1 + ipilimumab | 1 year PFS of 82% versus 39%, favouring patients with a favourable ctDNA profile; 1 year OS was 100% for patients with a partial response, 82% for those with a favourable ctDNA profile, and 39% for those with an unfavourable ctDNA profile |
| Seremet et al.43 | 85 patients with stage IV melanoma | mOS, mPFS and ORR; 21 months | Anti-PD1 | mPFS of 26 versus 9 weeks and mOS not reached versus 21.3 weeks favouring those with baseline ctDNA negative; HR for death was lower in those who became ctDNA negative (HR: 0.16) |
Clinical outcomes in locally advanced and metastatic melanoma with ctDNA-negative status at baseline or achieving ctDNA clearance following systemic therapy initiation, compared with those with persistent ctDNA positivity.
ctDNA = circulating tumour DNA; DMFS = distant metastasis-free survival; HR = hazard ratio; mOS = median overall survival; mPFS = median progression-free survival; ORR = overall response rate; OS = overall survival; PD-1 = programmed cell death protein 1; RFS = recurrence-free survival.
Finally, the predictive value of ctDNA analyses beyond the first-line treatment setting remains poorly understood. Marsavela et al. conducted a cohort study involving 125 plasma samples from patients with unresectable stage III and stage IV melanoma. While the study corroborated that absent or low baseline ctDNA levels are associated with longer PFS in the first-line setting, it failed to demonstrate a similar prognostic benefit in patients receiving second-line therapies, particularly those who had progressed following first-line targeted therapy. This limitation may be attributed to the cohort’s high proportion of patients with brain metastases, which are known to shed less ctDNA into the bloodstream.45
UM is the most common intraocular malignancy in adults and the second most common melanoma type.46 UM provides an intriguing platform for ctDNA detection experiments as it consistently harbours clonal, hotspot mutations, mainly affecting GNAQ, GNA11, PLCB4 and CYSLTR2. Other alterations, such as BAP1, SF3B1 and EIF1AX, are also useful in defining prognosis as they are associated with early (BAP1), late (SF3B1) or no (EIF1AX) metastasis risk.47 LB is particularly advantageous in UM due to its haematogenous spread pattern, the high tumour-derived molecular content in aqueous humour and the risks/complications associated with serial intraocular tissue sampling. Additionally, LB can aid in distinguishing UM from benign pigmented lesions (e.g. nevi) despite shared driver mutations (e.g. GNAQ/GNA11).48–50
Tebentafusp is the first drug to demonstrate clinically meaningful OS benefit in patients with metastatic UM. Exploratory analyses from the phase II IMCgp100-102 (Clinical and Molecular Response to Tebentafusp in Previously Treated Patients with Metastatic Uveal Melanoma: A Phase 2 Trial; ClinicalTrials.gov identifier: NCT02570308) and phase III IMCgp100-202 (Three-Year Overall Survival with Tebentafusp in Metastatic Uveal Melanoma; ClinicalTrials.gov identifier: NCT03070392) clinical trials hypothesized that ctDNA could help predict early treatment response.51–53 This would be particularly relevant for patients with progressive disease at first assessment, given that patients with lower ctDNA levels derived clinical benefit from tebentafusp compared with those receiving standard treatment.52,53 A prospective analysis of 69 patients with metastatic UM treated with tebentafusp assessed ctDNA dynamics using droplet digital polymerase chain reaction (PCR). In this study, ctDNA detection before treatment, found in 61% of patients, correlated with poorer prognosis and shorter OS (12.9 versus 40.5 months for those with undetectable ctDNA; p<0.001). In contrast, those who achieved a 90% or greater reduction in ctDNA at 12 weeks experienced significantly prolonged survival (median: 21.2 versus 12.9 months; p=0.02).54 Carvajal et al., in a phase II study, not only confirmed the prognostic role of ctDNA levels and ctDNA clearance throughout treatment but also validated this tool to differentiate responders from non-responders in patients with radiologically stable disease at their first assessment. This is particularly relevant given the clinical and radiologic dissociation, a common occurrence in patients with UM.55
Challenges and uncertainties
The implementation of ctDNA analysis faces critical challenges in low- and middle-income countries (LMICs), including unequal access to next-generation sequencing, inadequate infrastructure, funding constraints and shortages of trained personnel.56 Technical limitations, such as insufficient sensitivity/specificity for MRD detection and early cancer screening, alongside low ctDNA concentrations in low-shedding tumours, further hinder clinical utility.57 The lack of large-scale validation studies in diverse populations undermines confidence in ctDNA’s predictive value (e.g. negative results cannot exclude residual disease, and early recurrence detection lacks proven mortality benefit). Logistical hurdles, such as tumour tissue requirements for personalized assays and variable efficacy of non-personalized approaches (e.g. methylation markers), compound these issues. Improved clinical validation, deeper sequencing investments and equitable access to advanced technologies are essential to integrate ctDNA testing into routine oncology care in LMICs.57–59
Beyond financial and technological barriers, logistical challenges hinder personalized ctDNA testing in resource-limited settings. Personalized assays often require tumour tissue and advanced sequencing platforms, which are frequently unavailable. Non-personalized approaches (e.g. targeting common mutations or methylation markers) provide broader applicability but show inconsistent efficacy across malignancies.60 Detection sensitivity is further limited by low ctDNA concentrations in peripheral blood, particularly in low-shedding tumours, where few ctDNA molecules carry actionable mutations. Improving detection may necessitate expanded mutation panels or methylation targets, requiring deeper sequencing and greater costs.59 Ultimately, routine ctDNA integration in LMICs demands large-scale clinical validation across diverse cohorts and equitable access to high-quality sequencing technologies.
Conclusion
In conclusion, melanoma still exhibits critical gaps, such as defining the biological risk for thin tumours, reducing overtreatment in the adjuvant setting, establishing evidence-based durations for systemic therapy and optimizing surveillance strategies. Although LB is a promising tool, the current body of evidence remains largely preliminary and under development, preventing its broad and uncritical application in clinical practice. To date, the most compelling data support its use in assessing MRD in advanced stages and in response assessment in UM. The authors believe that accumulating data from a larger number of trials is crucial for a better understanding, particularly concerning the role of other biomarkers in this context. Such questions will be more definitively addressed through larger trials, guided by scientific evidence and recommendations from leading societies, thereby contributing to the refinement of personalized medicine. Barriers may include costs and access to the necessary technology for testing. These efforts will be essential in driving the field forward towards more individualized, effective and durable treatment strategies for melanoma.