Acute myeloid leukaemia (AML) is a complex and heterogeneous disease characterised by a multitude of molecular abnormalities. Better understanding of the mutational landscape has resulted in the development of targeted treatments in the last decade.1,2 AML is the most common myeloid leukaemia in adults; the latest age-adjusted incidence rate of AML in the United States and Europe was 4.3 and 3.7 cases per 100,000, respectively.3,4 The European LeukemiaNet (ELN) risk classification recognises patients with good, intermediate and adverse risk profile.5 Overall, the 5-year survival following a diagnosis of AML is around 20% across all risk categories.1 However, 5-year survival following diagnosis is around 40% in patients aged 25–64 years but is only around 5% in those aged ≥65 years.1 Since AML primarily affects older adults, this accounts for a huge number of years of life lost and represents a considerable therapeutic challenge.6
Treatment has not much changed in the last decades. Standard induction therapy for AML in patients tolerating intense treatment is typically a combination of an anthracycline (such as daunorubicin, idarabucin or mitoxantrone) and cytarabine (3+7), and sometimes the addition of etoposide.5,7,8 For patients not fit for intensive therapy, treatment should be individualised with patient and disease-specific characteristics taken into account. In elderly patients unsuitable for intensive therapy, either decitabine, azacytidine or low-dose cytarabine could be used as initial therapy.9–11 Despite intensive induction therapy, 10–40% of newly diagnosed patients with AML do not achieve a complete response and are, therefore, characterised as primary refractory.10 After achieving complete response, intermediate and adverse-risk patients with AML are usually consolidated with an allogeneic stem cell transplantation (alloSCT).10,12 The latter is not commonly used in elderly patients or in those with significant comorbidities.6,13 Although some improvement in outcome has been achieved in the last decades, there is a high medical need to improve the outcomes for patients with AML.
AML is a heterogeneous malignancy as it can be driven by many genetic and epigenetic lesions.14 Midostaurin, a multitargeted tyrosine kinase inhibitor whose targets include fms-related tyrosine kinase 3 (FLT3) and the KIT mutation, has demonstrated substantial clinical activity as a single agent or in combination with chemotherapy in patients with AML.15 Midostaurin has received regulatory approval for the treatment of newly diagnosed AML that is FLT3 mutation-positive, in combination with intensive standard chemotherapy,16 following the findings of the RATIFY phase III study.15,17 Midostaurin is the first oral FLT3 inhibitor to offer an extended survival for patients with FLT3-mutated AML. As a result, midostaurin is likely to become the standard of care for patients with mutations in the FLT3 and KIT genes. This article aims to provide an overview of the use of midostaurin in AML, as well as addressing some of the practical aspects associated with its use, including management of adverse effects and the potential for drug–drug interactions.
Overview of the use of midostaurin in acute myeloid leukaemia
FLT3 is expressed in more than 80% of AML samples. Length mutations resulting from internal tandem duplication (ITD), which are associated with an adverse prognosis, are expressed in 20–25% of adult AML patients.18–20 AML patients with FLT3 ITD mutations have significantly poorer overall survival (OS) and disease-free survival, as compared with patients with wild-type FLT3.18 Higher rates of relapse and poor OS are largely dependent on the FLT3 ITD to wild-type allelic ratio. The ELN AML 2017 recommendations also take this ratio into account in order to assess the risk profile. The prognostic impact of FLT3 ITD is also affected by concurrent mutations, such as nucleophosmin 1.21 Also, other co-occurring mutations, for example in DMT3A, are changing the outcome of FLT3 ITD-mutated AMLs. Relapse during AML treatment can be associated with an increase in the FLT3 mutant allelic ratio frequency, which may result in resistance to FLT3 inhibitors such as midostaurin.22 In addition to FLT3 ITD mutations, mutations in the tyrosine kinase domain (TKD) have been described in AML. FLT3 TKD mutations are present in 5–8% of patients with AML,19,23 but, unlike FLT3 ITD mutations, appear to have little or no significant impact on disease prognosis.23
The addition of midostaurin to standard chemotherapy (induction and consolidation), plus 12 cycles as a single-agent maintenance therapy, has been shown to significantly prolong OS and event-free survival among patients with AML and an FLT3 mutation, independent of allelic ratio.15 In RATIFY, compared with patients receiving standard chemotherapy plus placebo, those receiving chemotherapy plus midostaurin had significantly longer OS (74.7 versus 25.6 months, hazard ratio [HR] for death: 0.78; p=0.009) and event-free survival (8.2 versus 3.0 months, HR for event or death: 0.78; p=0.002).15 In a recent post-hoc analysis, midostaurin reduced the cumulative incidence of relapse compared with placebo, irrespective of the definition of complete remission in analyses where transplant was not accounted for. When transplant was treated as a competing risk there was not a meaningful difference between the treatment arms, suggesting that transplant in complete response was also important in preventing relapse.17 Another recent post-hoc subset analysis of the RATIFY data examined the contribution of maintenance therapy with midostaurin to overall outcomes and found that midostaurin was well tolerated, but could not provide definitive conclusions on the impact of maintenance therapy. In part, this was due to over half of midostaurin-treated patients receiving alloSCT and therefore, as per protocol, not receiving maintenance therapy.24However, the RADIUS study investigated the addition of midostaurin to standard of care in patients with AML and FLT3 ITD mutations after alloSCT, and found a 46% relative reduction in the risk of relapse with the addition of midostaurin at 18 months.25 Similarly, an interim analysis of the AMLSG 16-10 study reported the addition of midostaurin as maintenance therapy after alloSCT or high-dose cytarabine to be feasible and effective.26
The optimum use of midostaurin in AML remains an active area of research. Numerous questions remain unanswered, including its use in combined treatment with other regimens (such as other anti-methylating agents, other anthracyclines and new drugs) and its use in other settings (e.g. after allogeneic haematopoietic stem cell transplantation [alloHSCT], core binding factor AML or non-mutated FLT3). A number of clinical trials are attempting to answer these questions. Current and upcoming investigations include a study on the use of midostaurin in patients with AML whose FLT3 mutant to wild type signal ratio is below the 0.05 clinical cut-off (ClinicalTrials.gov Identifier: NCT03512197);27 an assessment on the use of midostaurin in paediatric populations (ClinicalTrials.gov Identifier: NCT03591510);28 further investigation into the efficacy and safety of midostaurin as a maintenance therapy in FLT3-mutated AML (ClinicalTrials.gov Identifier: NCT03280030);29 and assessment of variations on the standard midostaurin and chemotherapy regimen used in the RATIFY study (ClinicalTrials.gov Identifier: NCT03379727).30 Further optimisation of the standard chemotherapy regimen that midostaurin accompanies may be possible: in the RATIFY study, patients received daunorubicin at a dose of 60 mg/m2 body-surface area per day, but there is evidence that a higher dose of 90 mg/m2 may have additional benefits, particularly in those patients with FLT3 ITD mutations.6,31–33 Finally, ongoing real-world use will establish the long-term safety profile of midostaurin in AML, and clarify the currently unknown risk of off-target effects with long-term use of a multi-kinase inhibitor. Stroke is a theoretical concern; indeed, there have been reports that vascular endothelial growth factor (VEGF)-R2 inhibitors can produce unwanted side effects, such as intracranial bleeds in humans,34,35 and can worsen ischemic injury in neonatal rats.36 Fortunately, to date, stroke has not been reported in various studies investigating multi-kinase inhibitors.
Optimising midostaurin use
Prompt molecular testing for FLT3 ITD and TKD mutations is essential to initiate midostaurin as described in the product label.10,19 Every effort should be made to ensure fast turnaround times to achieve optimal results16,37 – the ELN currently recommends a turnaround time of
48–72 hours for FLT3 tests and testing for both ITD and TKD mutations.6 Molecular testing for FLT3 in parallel with cytogenetic testing could reduce the turnaround time for molecular diagnosis and ensure that all newly diagnosed patients receive an FLT3 test versus only cytogenetically normal patients.38 In RATIFY, patients were diagnosed on day 0 and started therapy with midostaurin on day 8.15 Therefore, a turnaround time for results to be received by the physician of 7 days or less is essential to optimise treatment benefit. Every effort should be made to stick to the dosing schedule in RATIFY and start midostaurin on day 8.15,16,37 Therefore, in cases when test results are not available by day 7, midostaurin should be started as soon as possible when FLT3 testing results are available and continued for 14 days of treatment up to 48 hours prior to the next cycle of chemotherapy.15 The standard induction regimen consists of 7 days of cytarabine plus 3 days of daunorubicin. The label recommends starting midostaurin on day 8 (i.e., one day after the last dose of cytarabine) of the first induction cycle and continuing until day 21 (Figure 1).37However, some countries/regions regularly use other induction regimens.39–41
In a prior phase Ib (A2106) study, concomitant administration of midostaurin and chemotherapy had a worse safety profile than sequential administration of midostaurin after chemotherapy.42 In addition, the mean plasma concentration of daunorubicin 24 hours after the first dose was greater with concomitant midostaurin, suggesting the possibility of a pharmacokinetic interaction between the two compounds.42 Based on these results, in RATIFY, midostaurin was started 1 day after the end of chemotherapy (cytarabine) to avoid or limit overlapping toxicity. Midostaurin was stopped 1 week prior to the next chemotherapy cycle (either induction 2 or consolidation) to facilitate blood count recovery.15
Based on these data, the following dosing schedule for midostaurin might be appropriate for patients receiving alternative chemotherapy regimens. In some regions, 10+3 chemotherapy (10 days of cytarabine plus 3 days of an anthracycline) is commonly used.39,40 While the dosing schedule in RATIFY should be adhered to as closely as possible,15,16,37 for those patients receiving anthracycline through day 10, at the discretion of the treating physician, midostaurin can be started on day 11 and given for 14 days up to day 24 of that cycle. In some institutions, the 5+2 regimen
(5 days of cytarabine plus 2 days of an anthracycline) is preferred.43 In these instances, it is recommended to use midostaurin on days 8 through 21 (14 days of treatment). If midostaurin is to be discontinued, the dose can be tapered or completely withdrawn, as preferred by the clinician, without the risk of a rebound effect.

Pharmacokinetics and pharmacodynamics of midostaurin
Midostaurin is administered with food at a dose of 50 mg twice daily in patients with FLT3 mutation-positive AML or 100 mg twice daily in patients with adult patients with aggressive systemic mastocytosis, systemic mastocytosis with associated haematological neoplasm, or mast cell leukaemia.16 The drug is rapidly absorbed after oral administration, with peak plasma concentrations observed at 1–3 hours following dosing (Table 1).16 After a radiolabelled midostaurin dose, the major circulating components in healthy volunteers were midostaurin (22.0%), and two active metabolites: CGP52421 (32.7%) and CGP62221 (27.7%).44 The plasma concentrations of midostaurin and CGP62221 accumulate in a time-linear manner in the first 3–6 days of daily oral dosing. Subsequently, the pharmacokinetics become nonlinear, with a large increase in bioavailability between day 5 and day 28 to reach a new steady state.45 Conversely, the longer-lasting metabolite, CGP52421, continues to accumulate to reach approximately seven times the concentration of midostaurin and CGP62221 at steady state.46,47 The metabolite CGP52421 may contribute a significant portion of the
anti-leukaemic activity observed in patients receiving oral midostaurin.48 A study in healthy volunteers found a high tissue distribution of the drug.46 It is not known whether midostaurin can cross the blood–brain barrier in humans; however, in an investigation of the tissue distribution using quantitative whole-body autoradiography in rats after an intravenous or oral administration of 14C midostaurin, radioactivity from 14C midostaurin was taken up by the pituitary gland and crossed the blood–brain barrier. The highest 14C concentrations were seen in the frontal cortex.49
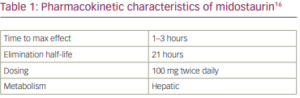
Trough plasma concentrations (Cmin) of midostaurin and its metabolites reached stable levels after one cycle (28 days) of treatment in all dosing regimens.50 Midostaurin and its metabolites have a long plasma half-life that ranges from 20.3 hours for midostaurin, 495.0 hours for CGP52421, and 33.4 hours for CGP62221, based on studies in healthy volunteers.44 Faecal excretion is the major pathway for the elimination of midostaurin, suggesting mainly hepatic metabolism, with minor renal clearance (4%).44 There is no clinically significant prolongation of the corrected QT (QTc) interval or relationship between changes in QTc and concentrations for midostaurin and its active metabolites.16 To avoid or limit overlapping toxicity with chemotherapy, it is therefore important to ensure that there are at least 48 hours between stopping midostaurin and starting the next cycle of chemotherapy. The prolonged half-life of midostaurin and its metabolites have prompted investigations of alternative dosing strategies, including administering midostaurin in formulations to circumvent plasma protein binding (e.g. gold nanoparticles), and high-dose pulse dosing as an alternative to prolonged daily dosing.51,52
Plasma inhibitory activity is a useful surrogate assay for monitoring the efficacy of FLT3 inhibition in patients treated with oral FLT3 inhibitors. Midostaurin has been shown to inhibit FLT3 activity in tumour cells overexpressing FLT3 ITD (IC50, <10 nM) and FLT3 TKD (IC50, <10 nM), as well as in cells expressing endogenous levels of wild-type FLT3 (IC50, 528 nM).53 In cultured AML blast progenitor cells expressing FLT3 ITD, midostaurin combined with decitabine was shown to result in greater levels of apoptosis than either agent alone.54
The efficacy of midostaurin in FLT3-driven myeloid disease was tested in a mouse model of FLT3 ITD-induced myeloproliferative disorder. Administration of midostaurin prolonged survival and decreased white blood cell count, myeloid hyperplasia, and spleen weight in mice transplanted with haematopoietic stem cells expressing FLT3 ITD.53 Midostaurin is highly selective in terms of plasma inhibitory activity for FLT3, whereas CGP52421 is less selective and more cytotoxic against primary blast samples in vitro. This suggests that non-selectivity may constitute an important component of the cytotoxic effect of FLT3 inhibitors in FLT3-mutant AML.48
Few renal or hepatic toxicities have been reported in clinical trials of midostaurin, although this might reflect the fact that eligibility criteria required adequate baseline renal and hepatic function. A current study is investigating the pharmacokinetics and safety of midostaurin in subjects with impaired hepatic function (ClinicalTrials.gov Identifier: NCT01429337).55
Drug–drug interactions and their impact on the pharmacokinetics of midostaurin
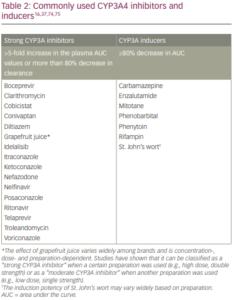
Cytochrome P450 (CYP3A4) is a major source of variability in drug pharmacokinetics and response.56 CYP3A4 is involved in the hepatic clearance of midostaurin, CGP52421 and CGP62221.44 Midostaurin, CGP52421 and CGP62221 are both inhibitors and inducers for CYP3A and, therefore, have the potential for drug–drug interactions with CYP3A4 modulators/CYP3A substrates.44
In an analysis from the RATIFY trial, the CYP3A4 inhibitors administered most frequently were posaconazole and voriconazole.57 Strong CYP3A4 inhibitors were used in 60.8%, 45.6% and 10.8% of patients during induction, consolidation and maintenance, respectively. This resulted in a 1.44-fold increase in midostaurin exposure (Cmin) compared with patients not receiving strong CYP3A4 inhibitors (n=55 versus n=112, respectively). No effect was observed on levels of CGP62221 and CGP52421. This increase in midostaurin exposure was not associated with a notable increase in the rate of midostaurin-related adverse events. Exposure-safety analyses showed that the time to occurrence of grade 3 or 4 clinically notable adverse events was slightly shorter with increasing exposure, but this was considered acceptable. In addition, exposure-response analyses found that a higher dose intensity was associated with greater effectiveness, in terms of complete response, event-free survival and OS. The investigators concluded that, although co-administration of midostaurin with strong CYP3A4 inhibitors without midostaurin dose adjustment, led to a balanced safety and efficacy profile in the RATIFY study, alternative medicinal products that do not strongly inhibit CYP3A4 activity should be considered.57
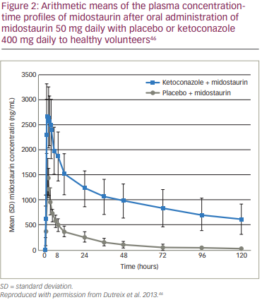
A drug interaction study showed that co-administration of multiple doses of midostaurin (100 mg twice daily on days 1–2 and 50 mg twice daily on days 3–28) with itraconazole 100 mg given twice daily on days 22–28 for 13 doses increased Cmin concentrations of midostaurin, CGP62221 and CGP52421 on day 28 by 2.1-fold, 1.2-fold and 1.3-fold, respectively, compared with the respective day 21 Cmin concentrations with midostaurin alone.16 In three phase I studies of healthy volunteers, co-administration of the potent CYP3A4 inhibitor ketoconazole with midostaurin resulted in a ten-fold increase in midostaurin exposure (Figure 2), while induction of CYP3A4 by rifampicin decreased midostaurin exposure by more than ten-fold.46 Midostaurin, administered as a single dose or in multiple doses, did not appear to affect the concentrations of midazolam or its metabolite 1′-hydroxymidazolam. It may therefore be concluded that midostaurin does not appear to inhibit or induce CYP3A4 in vivo under current clinically relevant conditions. However, this study was unable to reach a conclusion for CGP62221 and CGP52421 because of their low exposure following a single dose or 4–5 days of daily midostaurin dosing.46 In another study, a dose reduction to 12.5% of the initial dose of midostaurin in case of co-medication with strong CYP3A4 inhibitors
(e.g., posaconazole) did not affect response or outcome.26
The US and European labels for midostaurin recommend considering alternative therapies that do not strongly inhibit CYP3A4 or monitoring for increased risk of adverse reactions. Alternatively, if co-administered with strong CYP3A inhibitors, monitor patients for increased risk of adverse reactions, especially during the first week of administration in each cycle of chemotherapy in patients with AML and avoid use of CYP3A4 inducers (Table 2).16,37 The product label for midostaurin does not give specific advice on prophylactic antibiotics/antifungals or dose reductions,16because their impact was considered minimal. However, dose reduction should be considered in case of toxicity. In addition, grapefruit is a CYP3A inhibitor and, as such, should not be consumed with midostaurin.16
Midostaurin, CGP52421 and CGP62221 inhibit CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP2E1, and induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 and CYP3A in vitro.16 Further study is warranted regarding potential interactions of midostaurin with these cytochrome inhibitors/inducers. No other drug-drug interactions have been reported, although the US labelling instructions for midostaurin recommend interval assessments of QT by electrocardiogram if midostaurin is taken concurrently with medications that can prolong the QT interval.16
How drug–drug interactions may impact patients taking midostaurin
Invasive fungal disease is a major complication in AML.58 Fluconazole has shown efficacy in preventing invasive candidiasis during AML remission induction therapy. However, fluconazole lacks activity against moulds, and resistance among Candida species to fluconazole is increasing. Instead, itraconazole, voriconazole and posaconazole are used for prophylaxis against infections caused by Candida species and moulds.59
Posaconazole is the most widely used agent for prophylaxis in AML.60 It has been shown to give a significant survival and outcome benefit in prophylaxis during AML induction therapy.61 However, posaconazole is a strong CYP3A4 inhibitor, so, despite evidence showing that it does not impact the safety profile of midostaurin,57 should ideally be avoided with midostaurin.59 Similarly, other azole antifungal agents affect CYP3A4 enzymatic activity and, hence, should not be used during midostaurin therapy.62 In an ongoing clinical trial (ClinicalTrials.gov Identifier: NCT02634827), the use of all azoles except fluconazole is discouraged in patients requiring treatment with antifungal antibiotics.63 However, in the case of proven or suspected invasive fungal infections, agents such as posaconazole may be necessary. Temporary cessation or dose reduction of midostaurin is one option in such instances, but in light of the evidence from the RATIFY study showing no notable increase in midostaurin-related adverse events in patients treated with strong CYP3A4 inhibitors (including posaconazole) compared with those not receiving CYP3A4 inhibitors,57 the recommended course of action is to continue midostaurin without dose modification whilst closely monitoring the patient for toxicity.
Consensus general guidelines on the use of antifungals suggest minimising the risks of drug–drug interactions by clinical assessment, laboratory monitoring, avoidance of particular drug combinations, dose modification, optimal timing of oral drug administration in relation to meals, and use of pre-hydration and electrolyte supplementation if required.64 In addition, alternative agents for antifungal prophylaxis are being developed. For example, echinocandins, the newest class of antifungal agents, are poor substrates for CYP3A4 and include anidulafungin, caspofungin and micafungin.65 However, these novel antifungals are expensive and are not typically used as prophylaxis.66
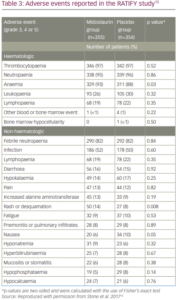
Management of adverse events during midostaurin treatment
Midostaurin has shown good tolerability among patients in the completed clinical trials to date.15,16 However, adverse reactions led to dose modifications (interruption or reduction) in 56% of patients and treatment discontinuation in 21%,16 and higher doses (100 mg twice daily) are associated with higher rates of discontinuation than lower doses (50 mg twice daily).42 The most frequent adverse reactions (>5%) were gastrointestinal symptoms, QT prolongation, neutropenia, pyrexia, thrombocytopenia, gastrointestinal haemorrhage, lipase increase and fatigue (Table 3).15The adverse event profile may be altered when midostaurin is used in combination with agents other than conventional cytarabine and daunorubicin; for example, in combination with azacitidine or decitabine, rates of gastrointestinal and pulmonary toxicities may be increased.67
Awareness of these potential toxicities and regular monitoring is important.16 The European Medicines Agency (EMA) product label recommends dose interruptions in the case of grade 3/4 pulmonary infiltrates, other grade 3/4 non-haematological toxicities, QTc interval >500 ms, grade 4 neutropenia (absolute neutrophil count <0.5 x 109/l) and persistent grade 1/2 toxicity. A dose reduction to 50 mg once daily is also recommended if the QTc interval exceeds 470 ms but is ≤500 ms.37
Nausea has been reported in more than 80% of cases during treatment with midostaurin, although grade ≥3 nausea was reported in only a minority of patients (6%),16 although the effects of persistent grade 2 nausea on patient quality of life should not be underestimated. As a result, the midostaurin labelling indications recommend administration of prophylactic antiemetics before treatment to decrease the risks of nausea and vomiting.16 In clinical studies, nausea and vomiting were generally managed with antiemetics. There are no specific recommendations on antiemetics to use alongside midostaurin although guidelines for antiemetic use are available in Europe and the US.68,69 If grade 3–4 nausea and/or vomiting occurs despite optimal antiemetic therapy, midostaurin dosing should be interrupted for 3 days (6 doses), then resumed at 50 mg twice daily; if tolerated, it can then be increased to 100 mg twice daily.16
In addition to nausea and vomiting, rash has been reported in up to 14% of patients receiving midostaurin (grade ≥3 in 3%).16 No specific information is given on the product label for the management of rash; however, a number of organisations, including the American Academy of Dermatology and the European Dermatology Forum, provide guidelines on the management of skin conditions.70,71
High-risk patient groups and midostaurin treatment
Midostaurin is well tolerated across a range of patient groups, including high-risk AML patients.69,72,73 Clinical studies have reported no impact of age on response and outcomes.26 A study of midostaurin plus azacitidine in elderly patients (median age 73 years) showed that 75 mg orally, twice daily increased trough levels of midostaurin during cycle 2 compared with cycle 1, with persistent and increasing levels of the active metabolite CGP52421. However, the combination was generally well tolerated.72 The addition of midostaurin to intensive induction therapy and as maintenance after alloHSCT or high dose cytarabine has been shown to be feasible and effective in a single-arm clinical study (n=147).73 The efficacy and safety of midostaurin has not yet been established in paediatric populations.16
Based on findings in animal studies, midostaurin may cause foetal harm when administered to pregnant women.16 In the absence of conclusive data, it is recommended to advise pregnant women of a potential risk. Since midostaurin has been found to pass into the milk of lactating rats, women are also advised not to breastfeed during treatment with midostaurin.16
Conclusions
Patients with AML harbouring the FLT3 mutation have an inferior OS and higher relapse rate compared with patients without a FLT3 mutation,18 and current treatment options are limited. The use of midostaurin in combination with standard cytarabine and daunorubicin induction, and cytarabine consolidation, has demonstrated improved OS in AML patients with the FLT3 mutation.15 Midostaurin is therefore likely to play a major role in the induction treatment of AML. However, a number of practical aspects should be considered when prescribing midostaurin.
Midostaurin is metabolised to the active metabolites CGP52421 and CGP62221, which exhibit prolonged cellular retention.44 Midostaurin, CGP52421 and CGP62221 are both inhibitors and inducers for CYP3A and, therefore, have the potential for drug–drug interactions with CYP3A4 modulators/CYP3A substrates.44 As a result, in situations where antifungals, including posaconazole and other azole antifungal agents are administered alongside midostaurin, patients should be closely monitored for midostaurin-related toxicity.16,37 While clinical data suggest that the coadministration of the antifungal medications posaconazole and voriconazole with midostaurin does not impact on its safety profile, caution is advised.57
To date, midostaurin has shown good tolerability among patients in the completed clinical trials. However, its use is associated with several adverse events, mostly gastrointestinal toxicity and rash.15,16 Although the risks of nausea and vomiting can be managed using prophylactic antiemetics before treatment,16 these adverse effects may be treatment-limiting in a minority of patients.
In conclusion, midostaurin represents an effective treatment for use in combination therapy in patients with newly diagnosed AML and a FLT3 mutation. The tolerability profile of midostaurin may facilitate its use in AML patients who cannot tolerate intensive induction therapy, in particular elderly people.