Congenital brain tumours are infrequent and account for approximately 1–4% of all paediatric cases.1 One to four live births per 100,000 are estimated to be affected by a congenital brain tumour.2,3 The majority of these neoplasms reside in the supratentorial compartment and, as noted by Volpe, the clinical manifestations of congenital brain tumours usually involve one or more of four typical syndromes.4 These syndromes are predicated on:
- macrocrania;
- hydrocephalus;
- focal neurologicsl deficit(s); and
- acute intracranial haemorrhage.
Although neuroimaging modalities are increasingly identifying cases during foetal development, aggressive surgical and chemo-/radiotherapeutic regimens have yet to significantly alter the dismal prognosis for most of these unfortunate patients.5 As determined by Isaacs in his 2002 review, overall survival is only 28%.2,3
Numerous small case series and larger reviews have been published on the topic of early-onset paediatric brain tumours.2,3,5–13 Ascertaining the frequency of individual congenital tumour types is often made difficult by the inclusion of older children in some analyses (up to 18 months of age in one study).11–13 In particular, these latter studies often underestimate the frequency of teratomas. For example, in Larouche et al.’s review of 1,289 infants less than one year of age, astrocytomas accounted for 30.5% of the cases, while teratomas accounted for only 4.9% of the total.11 Of the larger studies that were more strictly confined to the congenital time period, i.e. all cases less than or equal to two months of age, teratomas and astrocytomas were usually the most frequent tumour type.2,3,5,8 In Isaac’s 2002 analysis of 225 congenital brain tumours, teratomas (44.9%) were by far the most frequent type, followed by astrocytomas (16%) and medulloblastomas (7.6%).2,3 Buetow et al.’s 1990 review of 45 congenital cases included three main categories: 13 astrocytomas (nine grade I–III and four glioblastoma); 12 primitive neuroectodermal tumours (PNETs), four of which were medulloblastoma; and 12 teratomas.8 Finally, the study by Cassart et al. in 2008 of 27 foetal cases included 13 teratomas and four glial tumours (subtype unspecified) without any medulloblastomas/PNETs.5
In larger series of congenital brain tumours, many (with the exception of Buetow et al.) fail to adequately describe the subtype and grade of individual astrocytomas. However, some of the smaller
studies are exceptional in their detail. In documenting nine cases of congenital brain tumour, four of which were teratoma, Carstensen et al. subtyped their four cases of astrocytoma into two glioblastomas and two anaplastic astrocytomas.7 Similarly, in Lasky et al.’s report of 12 congenital brain tumours, five gliomas were noted using WHO grading, including one each of gliosarcoma (grade IV), desmoplastic infantile astrocytoma (grade I), anaplastic oligoastrocytoma (grade III), glioblastoma (grade IV) and anaplastic astrocytoma (grade III).6 Based on these data, all grades (I–IV) of astrocytoma, and many of the different subtypes, appear to be represented in the congenital period; this includes very rare instances of gliosarcoma.
Gliosarcoma – What Can Be Learned from Adult Cases?
Gliosarcoma, WHO grade IV, is considered a variant of glioblastoma that exhibits not only elements of high-grade astrocytoma, but also areas of malignant mesenchymal differentiation, i.e. sarcoma. Gliosarcomas comprise approximately 2–5% of all glioblastomas.14–16 Despite their distinctive bi-phasic histology, gliosarcomas carry the same dismal prognosis as conventional glioblastomas.
In their study of 29 gliosarcomas, Sarkar et al. demonstrated that most patients died within 1.5 years of surgery and adjuvant radiotherapy.14,17 Likewise, Meis et al. calculated a mean survival of only 8.3 months among 26 cases.15 Moreover, gliosarcomas share a propensity for the supratentorial white matter of adults (mean patient age 52.1 years).18 Notably, paediatric cases of gliosarcoma are uncommon and congenital cases of gliosarcoma are very rare.19–21
Pathologically, the areas of high-grade astrocytoma and sarcoma in gliosarcoma are variably intermixed. Most distinctive are those cases where each component, glial and mesenchymal, is clearly demarcated on microscopy. As with glioblastoma, the astrocytic areas are characterised by cytologic atypia, mitotic activity, microvascular proliferation and necrosis (classically ‘pseudopalisading’) (see Figures 1A–C and 2C). The astrocytic areas are immunohistochemically positive for the intermediate filament glial fibrillary acidic protein (GFAP) and are reticulin-poor, while the reverse is true for the sarcomatous component (see Figures 2A and 2B). Typically, the reticulin-rich mesenchymal areas are architecturally arranged in a fibrosarcomatous-like pattern containing fascicles of malignant spindle cells (see Figures 1D and 1E). Immunohistochemical analysis of these sarcomatous areas often further support differentiation along mesenchymal lines (see Figure 2C). For example, Ang et al. demonstrated desmin positivity in four of 31 gliosarcomas, while Hochwald et al. illustrated prominent desmin positivity in the spindled component of a congenital gliosarcoma.21,22 The presence of desmin, an intermediate filament present in skeletal, smooth and cardiac muscle, would constitute evidence of muscular differentiation, i.e. rhabdomyosarcomatous. In support of their immunohistochemical results, Hochwald et al. offered an ultrastructural suggestion of myofilaments.21 The sarcomatous component of gliosarcoma may also take on the appearance of malignant fibrous histiocytoma, chondrosarcoma or osteosarcoma.23 Rarely, epithelial areas may be seen, suggesting carciomatous differentiation.24
The controversy regarding the histogenesis of gliosarcomas was largely resolved with the advent and implementation of modern genetic techniques. Given the distinct and often well circumscribed areas of glial and sarcomatous morphology, early pathological accounts proposed the gliosarcomas represented a ‘collision’ tumour, i.e. two separate neoplasms.18 Moreover, the sarcomatous element was felt by some authors to arise from regions of proliferative vascular mesenchyma, i.e. microvascular proliferation.25,26 The collision tumour hypothesis of gliosarcomas has since been abandoned in favour of a monoclonal histogenesis wherein the sarcomatous element is derived through either metaplasia or ‘dedifferentiation’ of the glial component.27 Subsequently, Biernat et al. demonstrated identical TP53 mutations in both the glial and sarcomatous components of two gliosarcomas via single-strand conformational analysis and direct DNA sequencing.28 Using interphase cytogenetics, i.e. in situ hybridisation, Paulus et al. demonstrated monosomy for chromosomes 10 and 17 in both the glial and sarcomatous areas of a gliosarcoma, suggesting a monoclonal origin for both components.29
In 2000, Reis et al. performed a detailed genetic analysis of gliosarcomas, looking for mutations that are commonly seen in glioblastoma. The TP53 and PTEN genes were analysed by polymerase chain reaction (PCR) single-strand conformation polymorphism analysis and direct sequencing, while the p16, epidermal growth factor receptor (EGFR), CDK4 and MDM2 genes were studied via differential PCR. With the exception of a lack in amplification in EGFR, gliosarcomas demonstrated all of the genetic abnormalities seen in primary glioblastoma (TP53 mutation, 5/19; PTEN mutation, 7/19; homozygous p16 deletion, 7/19; CDK4 and MDM2 of amplification, 1/19). Moreover, identical mutations for PTEN (two), p16 (one), TP53 (one) and CDK4 (one) were identified in the glial and sarcomatous components of a subset of these gliosarcomas.30
Using duplex PCR and single-strand conformation polymorphism analysis, Actor et al. also demonstrated similar glioblastoma-like alterations in 38 gliosarcomas (amplification of CDK4, MDM2, EGFR and PDGFRA genes in 11, 8, 8 and 3%, respectively; TP53 mutation in24%; and PTEN mutation in 45%). In addition, Actor et al. studied eight gliosarcomas by comparative genomic hybridisation after microdissection and universal DNA amplification and demonstrated that both components shared over half of the chromosomal imbalances detected.31 Overall, not only do these genetic studies provide support for the inclusion of gliosarcomas as a variant of glioblastoma, but they argue for a monoclonal origin for the histologically distinctive glial and sarcomatous elements.
Congenital Gliosarcoma – A Rare Entity
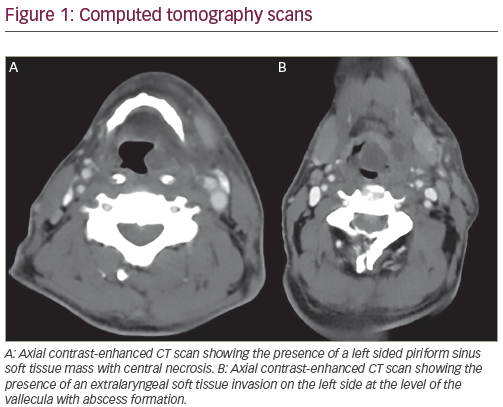
Only rare cases of congenital gliosarcoma have been documented in the literature. Among these, pathological and genetic details are generally quite limited. Although the first purported case of congenital gliosarcoma may well be attributed to Holt in 1917, Goldstein et al. appear to have reported the first case in the modern era.20,32,33 They described the case of a four-month-old female who presented at birth with macrocephaly and later developed lethargy. Computed tomography (CT) of the head revealed an enhancing cystic neoplasm replacing much of the left cerebral hemisphere. Peripheral calcification was noted. Based upon the radiological enhancement, Goldstein et al. interpreted this neoplasm to be malignant. A fairly detailed pathological description was offered and included spindle cells, rich reticulin deposition, calcification, mitoses, focal necrosis/ microvascular proliferation and a subset of cells with prominent hyaline cytoplasm (some of which were GFAP positive). Notably, many of those latter cells (depicted in their Figure 1D) contained prominent nucleoli.32 Unfortunately, surgical, treatment and follow-up details were not described.
Chadduck et al. published a subsequent instance of congenital gliosarcoma in a two-month-old female whose mother was exposed to the insecticide heptachlor via ingested dairy milk during pregnancy.34 This girl presented with projectile vomiting at two weeks of age and had steadily increasing macrocephaly. CT scanning revealed an enhancing lesion involving most of the right hemisphere, plus secondary hydrocephalus. Pathological details (reviewed by the renowned Dr L Rubinstein) were limited, but described spindled fibrosarcomatous-like cells and ‘endothelial proliferation’, i.e. microvascular proliferation. Cytogenetic analysis revealed a complex structural pattern, including evidence of two translocations, t(2;12)(q13;p11) and t(4;17)(q31;q25), in addition to the presence of three different chromosome markers that were not further characterised. After subtotal resection and monthly chemotherapy (including vincristine, BCNU, procarbazine, cytosine, arabinoside, cisplatinum and cytoxan) the child was free from tumour progression at 11 months.In detailing the radiological features of 12 neonatal brain tumours, Radkowski et al. included one case of congenital gliosarcoma affecting a 26-day-old male.35 CT scanning revealed patchy enhancement within a superficial right temporal lobe tumour. Magnetic resonance imaging (MRI) revealed hypointensity on T1, hyperintensity on T2 and minimal evidence of mass effect. Unfortunately, pathological details were not described. After gross total resection, the patient was alive at 21 months.
More recently, in 2000, Rizk et al. briefly described the presence of a right temporo-parieto-occipital gliosarcoma in a two-month-old infant born after in vitro fertilisation.36 This patient had macrocephaly from birth and on presentation had hypotonia and macrocephaly. MRI revealed an enhancing neoplasm, seemingly with a large cystic component, causing significant right-to-left midline shift. A gross total resection was achieved. Again, pathology was not detailed. Soon after surgery the patient developed diffuse intravascular coagulation and died three days post-surgery with significant bleeding.
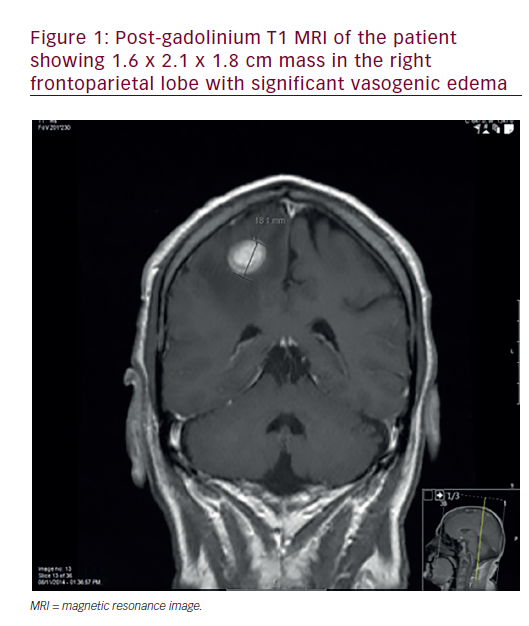
In the most detailed clinicopathological report to date, Hochwald et al. described the case of a newborn male who died as the result of a large left cerebral hemispheric gliosarcoma.21 CT and MRI revealed a solid and haemorrhagic mass causing significant mass effect and secondary hydrocephalus. The neonate expired at one day of age after withdrawal of care (and prior to any definitive tumour treatment). At autopsy, there was evidence of cerebral oedema and tonsillar herniation. The majority of the 10×8.5x5cm mass was haemorrhagic and necrotic, although a 3x2x5cm white portion of viable tumour was identified. Microscopically, this gliosarcoma was indistinguishable from those examples described in the adult literature. Morphologically, the astrocytic areas were glioblastomatous in morphology, typified by cellular atypia, numerous mitoses, microvascular proliferation and pseudopalisading necrosis. The prominent mesenchymal areas were reticulin-rich and populated by spindled cells arranged in a fibrosarcomatous pattern. In keeping with the strong desmin positivity in these latter areas, ultrastructural analysis suggested the presence of myofilamentous differentiation. Unfortunately, attempts at tumour analysis via array comparative genomic hybridisation were unsuccessful.
The pathological differential diagnosis of congenital gliosarcoma involves entities that are microscopically characterised by the presence of glial cells and a reticulin-rich spindle cell component that can at times mimic the appearance of sarcoma. Chief considerations include:
- desmoplastic tumours of infancy (DTIs), also known as desmoplastic infantile astrocytoma (DIA) and desmoplastic infantile ganglioglioma (DIG);
- ‘sarcoglioma’; and
- pleomorphic xanthoastrocytoma (PXA).
DTIs are WHO grade I neoplasms that are usually large and supratentorial. Neuroimaging further reveals enhancing solid and cystic components. Microscopically, the spindled mesenchymal component is accompanied by a variably conspicuous neuroepithelial component (including astrocytomatous and sometimes dysplastic neuronal cells), and at times a mitotically active small round blue cell-like component. Ultrastructural analysis reveals a basal lamina around the spindle cells. Gross total resection often results in long-term survival.37 PXAs are WHO grade II tumours that tend to affect older children and are often epileptogenic. These neoplasms are also predisposed to the superficial neocortex, especially the temporal lobe. Neuroimaging classically reveals a cystic neoplasm with an enhancing mural nodule. Microscopically, spindled cells are arranged in fascicles and are accompanied by lymphocytes, oeosinophilic granular bodies and large pleomorphic vacuolated cells. Once again, when viewed by electron microscopy, the spindled cells are often surrounded by basal laminae. Although originally often mistaken for glioblastoma, and then subsequently labelled as a WHO grade II tumour, recent studies have swung the pendulum back, suggesting that PXAs may in fact be more aggressive.38
Finally, in 1979 Lalitha and Rubinstein proposed the entity of ‘sarcoglioma’ to denote the presence of a primary intracranial sarcoma (intraparenchymal or meningeal based) that induces the presence of a more peripherally situated reactive glioma.39 Transitions from reactive to neoplastic (including glioblastoma) were purported in these secondary gliomatous areas. Given the clinicopathological features of these above three differential diagnoses, the legitimacy of possibly three previously reported cases of congenital gliosarcoma is drawn into question.
Goldstein et al. and Rizk et al. both illustrated images depicting very large and cystic tumours, findings that would be typical for DTIs and uncommon for gliosarcoma.18,32,36 Moreover, Goldstein et al. illustrated histology typified by cells containing abundant hyaline cytoplasm and seemingly prominent nucleoli, features that are reminiscent of the ganglioid cells of DTI, i.e. DIG. In addition, Radkowski et al.’s case of congenital gliosarcoma was situated in the superficial aspects of the right temporal lobe, a finding that is especially suspicious for DTI given the lack of associated mass effect around the tumour and the relatively long survival of 21 months.35 Since DTIs were not especially well recognised until their inclusion in the 1993 edition of the WHO classification of tumours of the central nervous system, it is not entirely unreasonable that the possibility of a DTI was overlooked in the differential diagnosis of the cases of Goldstein et al., Rizk et al. and Radkowski et al.18
Cases pertinent to the above differential diagnosis, and not previously recognised to be gliosarcomas, are also present in the literature. Among the eight original cases of so-called ‘sarcoglioma’ described by Lalitha and Rubinstein, three cases contained histological foci consistent with glioblastoma and fibrosarcoma; in particular, one of these three occurred in a 2.5-month-old male.39Although the concept of a secondary glioma arising out of a primary intracranial sarcoma is intriguing (i.e. sarcoma with low-grade glioma), its utility becomes moot in those cases with dual malignant features, i.e. sarcoma and glioblastoma, since it would be extremely difficult to determine which component was primary. Glioblastoma is exceedingly more common than primary intracranial sarcoma. Since genetic studies have proved that the sarcomatous element of gliosarcoma can share identical glioblastoma-like alterations with its malignant astrocytic counterpart, it would seem most appropriate to consider these rare cases of ‘sarcoglioma’ (i.e. with fibrosarcoma and secondary glioblastoma) as instances of gliosarcomas (i.e. primary gliomas with sarcomatous metaplasia/dedifferentiation).
With the exception of the cytogenetic results described by Chadduck et al. (see above), the genetics of congenital gliosarcomas is poorly understood.34 However, some insight into the genetics of congenital gliosarcomas may be drawn from the recent analysis of six congenital glioblastomas by Brat et al.1 Clinically, these congenital glioblastoma patients ranged from four days to 12 weeks of age. All tumours were supratentorial and all, save for one, involved the cerebral hemispheric lobes. Five of six were either biopsied or resected and two received chemotherapy. Surprisingly, three patients demonstrated extended survival (case 2, 67 months; case 3, 90 months; and case 4, 78 months). Also of note was the fact that these tumours infrequently exhibited the genetic defects characteristic of adult glioblastomas.
Fluorescence in situ hybridisation (FISH) analysis failed to reveal amplification of EGFR or 9p21 deletion, but demonstrated 10q deletion in two patients. In addition, sequencing of PTEN and TP53 did not uncover any mutations. As such, as gliosarcomas are considered a variant of glioblastoma, it would be reasonable to hypothesise that congenital gliosarcomas may not be characterised by the same genetic alterations that typify adult glioblastomas/gliosarcomas. However, this would not preclude congenital gliosarcomas from sharing genetic alterations with their gestational brethren congenital glioblastoma
.
Conclusion
Clearly, in-depth analysis of future cases of congenital gliosarcoma is required. Detailed pathological, genetic and treatment data are needed, especially given the encouraging results of Brat et al., which suggest that congenital glioblastomas are not only genetically different from their adult counterparts, but are also prognostically different given the extended survival seen in a subset of their patients (three patients with survival at or beyond 5.5 years).1 Hopefully, novel data will pave the road to an effective treatment for these unfortunate children.