Cholangiocarcinoma is an epithelial malignancy of the biliary system characterized by cholangiocyte differentiation, and its incidence is on the rise. Currently, cholangiocarcinomas are classified by their anatomical location in the biliary system as intrahepatic cholangiocarcinoma, distal cholangiocarcinoma, or hilar cholangiocarcinoma. Since 2010, the standard of care in all advanced, inoperable biliary tract cancers has been combination cisplatin plus gemcitabine, irrespective of anatomical subtype. Valle et al. demonstrated a median survival of 11.7 months with this combination compared with 8.1 months with single-agent gemcitabine in the phase III, randomized, controlled ABC-02 trial.1 Because of the known toxicity profile with combination chemotherapy, and the need for improved survival, drugs utilizing a targeted approach to treating cholangiocarcinoma have been pursued in recent years with the hope of providing more treatment options for these patients.
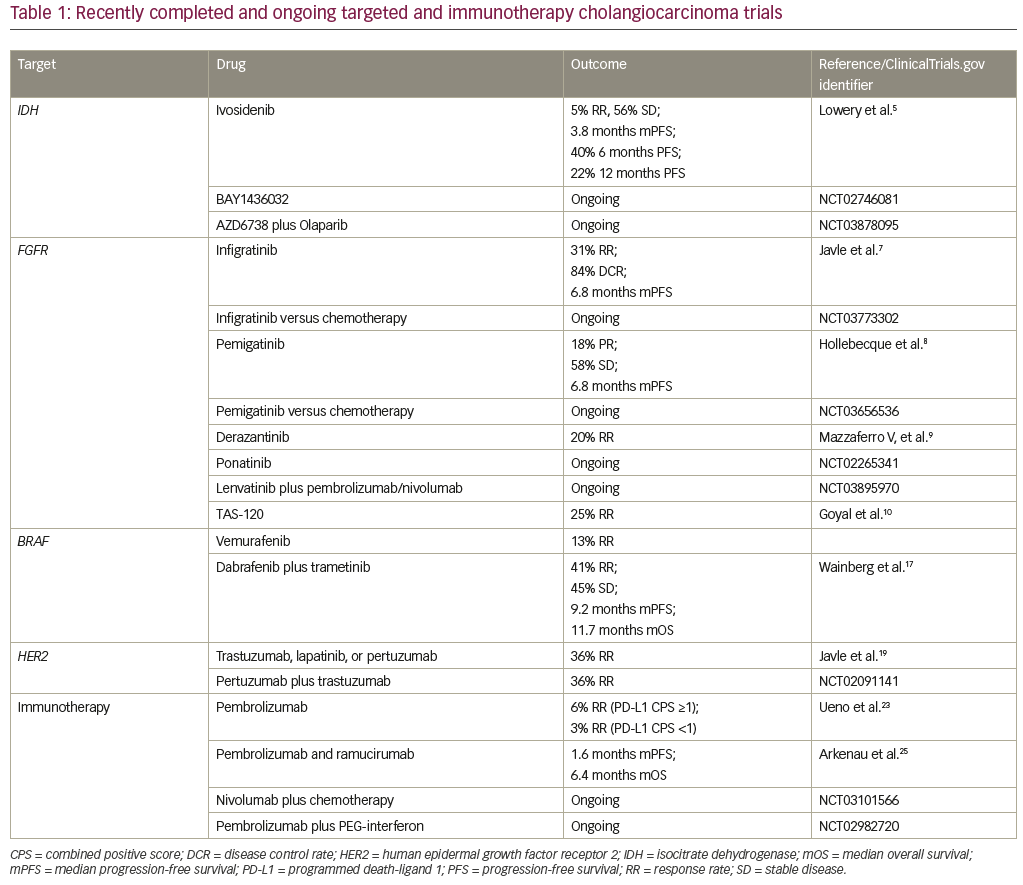
Precision oncology in cholangiocarcinoma has been facilitated by the elucidation of the genomic and epigenomic landscape of this disease by Jusakul et al. in an analysis performed by the International Cancer Genome Consortium.2 Four distinct etiological clusters were recognized, defined by differences in gene expression, mutation and copy number changes, and epigenetic changes. Cluster 1 was enriched with alterations in BRCA1/2 and ARID1A as well as human epidermal growth factor receptor 2 (HER2) amplification. Cluster 2 was enriched with TP53 alterations. Clusters 3 and 4 exhibited high copy number alterations and programmed cell death 1/programmed death-ligand 2 (PD1/PD-L2) expression and epigenetic mutations in isocitrate dehydrogenase 1/2 (IDH1/2), BRCA1 associated protein 1 (BAP1), and FGFR/PRKA-related gene rearrangements. Clusters 1 and 4 demonstrated distinct DNA hypermethylation patterns not seen in clusters 2 and 3. Anatomically, clusters 1 and 2 occur equally in intrahepatic and extrahepatic cholangiocarcinomas, with HER2 amplification occurring more commonly in distal tumors. Clusters 3 and 4 occur more commonly in intrahepatic cholangiocarcinoma. The characterization of these distinct molecular subtypes of cholangiocarcinomas highlights the need to better understand whether treatment of these patients should also be individualized and whether certain patients with cholangiocarcinomas may benefit from targeted and immunotherapies. Table 1 summarizes the targeted and immunotherapies discussed below.
IDH as a target
Whole-exome sequencing and integrative genomic analyses have identified recurrent mutational patterns involving IDH1 and IDH2 alterations in a subset of patients with cholangiocarcinoma.3,4 This alteration leads to the accumulation of the oncometabolite 2-hydroxyglutarate, which blocks cell differentiation, leading to the growth of undifferentiated tumor cells. It is estimated that up to 20% of intrahepatic tumors may harbor IDH mutations. As a result, numerous companies have developed IDH inhibitors, which are currently being tested in clinical trials.
Ivosidenib, AG-120, (ClinicalTrials.gov identifier: NCT02073994) is an oral, first-in-class, selective, reversible IDH1 inhibitor that has demonstrated a favorable safety profile in IDH1-mutated solid tumors.5 Data from a cohort of 73 heavily pre-treated patients with cholangiocarcinoma demonstrated a 5% response rate and stable disease in 56% of patients. The median progression-free survival was 3.8 months, the 6-month progression-free survival was 40%, and 12-month progression-free survival was 22%. Early biomarker data from paired biopsies obtained on trial suggests that mechanistically, ivosidenib restores differentiation of cells. Based on these encouraging findings, the phase III ClarIDHy trial (ClinicalTrials.gov identifier: NCT02989857) evaluated outcomes in patients receiving the drug compared to placebo with previously treated nonresectable or metastatic IDH1-mutated cholangiocarcinomas. Unique toxicities associated with ivosidenib, such as differentiation syndrome and liver toxicity, were identified in early phase trials with the drug and will also be better characterized in the ClarIDHy trial. BAY1436032 is another mutant IDH1-targeted small molecule in development. Mutations in IDH2 have been targeted in by the IDH2 inhibitor enasidenib in relapsed or refractory acute myeloid leukemia, which was recently approved by the US Food and Drug Administration (FDA) in this setting. The efficacy of targeting IDH2 in cholangiocarcinoma remains unclear. Given the mechanistic synergy between IDH inhibitors and PARP inhibitors, combination studies with these two classes of agents are also underway (ClinicalTrials.gov identifier: NCT03878095).
Fibroblast growth factor receptor as a target
FGFR alterations and fusions have been identified in a subset of patients with cholangiocarcinoma (cluster 4), resulting in aberrant activation of downstream receptor tyrosine kinase signaling.6 FGFR fusions are present in roughly 15% of intrahepatic cholangiocarcinomas and lead to ligand-independent activation of the FGFR pathway, which is felt to be a driver event.
The selective, ATP-competitive, oral pan-FGFR inhibitor BGJ398 (infigratinib) demonstrated favorable safety in a cohort of 61 heavily pretreated cholangiocarcinoma patients with FGFR amplification, alterations, or fusions.7 Early efficacy analyses revealed a 31% response rate and an 84% disease control rate in this cohort. It should be noted that 79% of the patients on this trial had FGFR fusions, and a minority had mutations and amplifications. The response rate in patients with FGFR fusions was 23% and there were no responders in patients with FGFR mutations. Median progression-free survival was 6.8 months. The front-line phase III study comparing BGJ398 to standard of care chemotherapy (gemcitabine plus cisplatin) for unresectable, metastatic cholangiocarcinomas harboring an FGFR2 gene fusions/translocation is ongoing (ClinicalTrials.gov identifier: NCT03773302). Similarly, INCB054828 (pemigatinib), a selective, potent oral inhibitor of FGFR1, 2, and 3, was evaluated in a phase III study
(ClinicalTrials.gov identifier: NCT02924376) of previously treated cholangiocarcinomas with FGFR2 translocations, FGF/FGFR genetic alterations, or no FGF/FGFR genetic alterations. Of 45 evaluable patients in the cohort with FGFR2 translocations, eight (18%) had a confirmed partial response and 26 (58%) had stable disease. Median PFS was 6.8 months. No responses were observed in cholangiocarcinomas with FGF/FGFR alterations.8 A front-line phase III study evaluating pemigatinib versus gemcitabine plus cisplatin chemotherapy in patients with cholangiocarcinoma harboring FGFR2 rearrangements, is also underway (ClinicalTrials.gov identifier: NCT03656536). These front-line trials will address the important, and intriguing, question of whether targeted therapies will be superior to chemotherapy in the front-line setting for subsets of cholangiocarcinomas, which has the potential to change standard of care.
Similarly, the orally available, pan-FGFR kinase inhibitor derazantinib (ARQ-070) was found to be safe and demonstrated a response rate of 20% in advanced intrahepatic cholangiocarcinoma with FGFR2 fusions.9 This drug has now moved into a phase III registration study. Additional early-phase studies with non-specific tyrosine kinase inhibitors such as ponatinib have also been tested in patients with advanced intrahepatic cholangiocarcinoma harboring an FGFR fusions with promising efficacy, and phase II studies are underway (ClinicalTrials.gov identifier: NCT02265341), as are combination studies with checkpoint inhibitors such as pembrolizumab (ClinicalTrials.gov
identifier: NCT03895970) in FGFR-altered solid tumors, including cholangiocarcinoma. The efficacy of these strategies remains to be seen.
The FGFR inhibitor TAS-120 is a covalent, pan-FGFR inhibitor that is highly selective and has demonstrated efficacy in patients with FGFR2 resistance mutations. It was found to have an objective response rate of 25% in patients with cholangiocarcinoma with FGF/FGFR aberrations, rearrangements, fusions, and amplifications.10 In this trial, seven of the 24 evaluable patients with FGFR fusions had partial responses, and an additional 15 had stable disease as best response. Twenty of the 24 patients were noted to have tumor regression. At the time of presentation, 15 patients with FGFR fusions were still on therapy. As was the case with BGJ398, patients with FGFR alterations did not have a considerable response rate to this drug, either. Interestingly, however, TAS-120 did demonstrate efficacy in four patients with FGFR2-fusion-positive intrahepatic cholangiocarcinoma who developed acquired resistance to the FGFR kinase inhibitors such as BGJ398 through the development of secondary FGFR2 kinase domain mutations.11 TAS-120 was granted orphan drug designation by the FDA in May 2018, and remains under study in the context of a global, phase II study for patients with cholangiocarcinomas harboring FGFR2 gene fusions.
It has now become imperative to further investigate mechanisms of acquired resistant to these drugs in order to improve on the durability of response seen in these patients. In preclinical studies and early clinical studies in urothelial carcinoma patients who were treated with BGJ398, the gatekeeper FGFR3 mutations in V443L, V443M, and L496V have been identified as mechanisms of acquired resistant. Prior biomarker analyses have also identified a polyclonal secondary FGFR2 kinase point mutation as a mechanism of acquired resistant to BGJ398. It remains to be seen whether these same mechanisms are responsible for resistance to the drug in cholangiocarcinoma and how this knowledge can be best leveraged for further drug development.
BRAF as a target
BRAF mutations have been reported in cholangiocarcinoma at a frequency of up to 5%.12 Transcriptomic profiling of surgically resected cholangiocarcinomas has suggested these mutations are associated with significantly worse prognosis.13 In the first basket study of BRAF-mutated non-melanoma cancers treated with BRAF inhibitor vemurafenib in 2015, it was noted that one of the eight BRAF-mutated cholangiocarcinomas experienced a partial response suggesting that it would be feasible to target this mutation in cholangiocarcinoma.14 Targeting BRAF with the combination of dabrafenib (BRAF inhibitor) plus trametinib (MEK inhibitor) has led to dramatic improvements in overall survival with subsequent FDA approval in BRAFV600E mutated melanomas and non-small cell lung cancer.15 Isolated case reports of responses to the combination of dabrafenib plus trametinib had previously been reported in BRAFV600E mutated cholangiocarcinomas.16 The first reports with the combination of dabrafenib (BRAF inhibitor) plus trametinib (MEK inhibitor) was presented by Wainberg et al., reporting outcomes from the biliary tract cohort of the phase II ROAR basket trial. The data revealed a 41% response rate in BRAFV600E mutated biliary tract cancers treated with dabrafenib and trametinib and stable disease as best response in another 45% of patients.17 The median progression-free survival in this study was 9.2 months and median overall survival was 11.7 months, which is comparable to outcomes with standard of care first-line chemotherapy. The cohort of patients consisted of mostly biliary tract adenocarcinoma (74%), with 17% hepatocholangiocarcinoma, and 9% with cholangiocarcinoma. Immunologic, cardiac, and ocular toxicity seen with this combination was consistent with that seen in prior trials treating other BRAF-mutated cancers. Moreover, biomarker analyses suggested that co-occurring overexpression of MAPK pathway genes were associated with resistance to the combination, highlighting the need for further research on which biliary tract cancer patients may best tolerate and most benefit from BRAF-targeted therapy.
Lessons from HER2
HER2/neu amplification has been estimated to occur in 12–15% of gallbladder and biliary tract cancers.18 HER2-directed monoclonal antibodies improve survival and are FDA approved for the treatment of advanced breast and gastric cancers. Moreover, HER2-directed small molecule tyrosine kinase inhibitors as well as antibody-drug conjugates are also FDA approved for HER2 amplified breast cancer. Recent studies have explored whether anti-HER2 directed therapy would be effective against HER2 amplified, overexpressed, or mutated biliary tract cancers.
In a study by Javle et al, nine patients with gallbladder cancer and five patients with cholangiocarcinoma were treated with HER2/neu-directed therapy (trastuzumab, lapatinib, or pertuzumab) in order to identify any potential signals for further investigation.19 In the gallbladder cohort, HER2/neu amplification was confirmed in eight out of nine cases and best response with HER2-directed therapy was noted to be a complete response in one patient, partial response in four patients, and stable disease in three patients. One patient with a HER2/neu mutation had a mixed response on lapatinib as best response. Interestingly, while the gallbladder cohort was composed mainly of HER2/neu-amplified tumors, the cholangiocarcinoma cohort was predominantly composed of patients with HER2/neu alterations; no responses were seen in the cholangiocarcinoma cohort. This highlights the relevance of distinguishing gene amplifications from gene alterations and may help to explain the variable responses to targeted therapy seen across anatomical locations in the biliary system. It serves as a reminder that potential driver mutations in one tumor type cannot be assumed to be so for biliary tract cancers and warrants testing in clinical trials. Preliminary data from 11 patients on the MyPathway study, testing dual HER2 blockade with pertuzumab plus trastuzumab in HER2-positive metastatic biliary cancers (eight HER2-amplified/overexpressed and three HER2 mutated), revealed partial responses in four patients (ClinicalTrials.gov identifier: NCT02091141). Accrual to this trial is ongoing; preliminary data suggest that dual HER2 blockade may serve as a promising therapeutic strategy in biliary tract cancers.
The case for immune checkpoint inhibitors
Transcriptomic and genomic sequencing data have identified a subgroup of cholangiocarcinomas with high tumor mutational burden, abundant tumor-specific neoantigens, and overexpression of immune-related genes, including genes encoding inhibitory immune-checkpoint proteins.6 Prognostically, this study identified this patient subgroup to have the worst survival. Moreover, tumor-associated macrophages, endothelial cells, and fibroblasts have been identified in the tumor stroma of cholangiocarcinoma and are thought to contribute to their immunosuppressive microenvironment, suggesting a possible role for immunotherapy in cholangiocarcinoma.20
Immune checkpoint inhibitors have revolutionized the treatment of solid tumors across histologies and have received FDA approval in both the adjuvant and metastatic settings in various cancer types. The use of immune checkpoint inhibitors in cholangiocarcinoma is currently being explored. Based on results from the KEYNOTE-028 study, the PD-1 inhibitor pembrolizumab is already approved for all refractory mismatch repair-deficient cancers, including cholangiocarcinoma. In the study, there was a 17% response rate in cholangiocarcinomas, with a median duration of response of 40 weeks with one patient with a complete response and four with stable disease as best response.21 This data prompted the KEYNOTE-158 basket trial. Preliminary data from 104 patients treated with pembrolizumab for advanced cholangiocarcinoma reported an overall response rate of 6% among those with PD-L1 combined positive score (CPS) ≥1 (n=61) and 3% among those with PD-L1 CPS <1 (n=34).22 Numerous other single-agent checkpoint inhibitors (ClinicalTrial.gov identifiers: NCT03110328, NCT02829918, and NCT01938612) and combination checkpoint inhibitor studies (ClinicalTrial.gov identifier: NCT03101566) remain in progress.
Mismatch repair deficiency has been reported in about 5% of cholangiocarcinomas, suggesting that immunotherapy-based therapy may be effective in a subset of all cholangiocarcinomas, although subtype-specific analyses remain to be completed. Other predictive biomarkers of response to immune checkpoint inhibitors remain under investigation, such as tumor mutational burden, which has been associated with response to immune checkpoint inhibitor therapy in non–small cell lung cancer, melanoma, and urothelial cancers. Next-generation sequencing studies have reported that roughly 5% of cholangiocarcinomas may harbor high tumor mutational burden.6 It remains to be seen whether tumor mutational burden will be validated as a biomarker of response in cholangiocarcinoma and whether this subset of patients may benefit from immune checkpoint inhibitor use. PD-L1 positivity, at varying thresholds, also continues to be explored as a biomarker for response to immune checkpoint inhibitor therapy across solid tumors. In perihilar and intrahepatic cholangiocarcinoma, PD-L1 expression has been detected in up to 60% of biopsies—both in the tumor and the tumor microenvironment.23 Further investigation is needed to better understand whether PD-L1 status can be applied as a predictive biomarker for response to checkpoint inhibitor therapy.
Combination studies with investigational microenvironment modulating agents have also been tested. A study combining pembrolizumab and ramucirumab (ClinicalTrial.gov identifier: NCT02443324) was found to be safe, but it did not appear to improve efficacy. One response was seen among 26 patients, and median progression-free and overall survival (1.6 and 6.4 months, respectively) did not seem to be better than historical control.24 The addition of GM-CSF to pembrolizumab (ClinicalTrial.gov identifier: NCT02703714) has been found to be safe and partial responses have been reported in five out of 24 (21%) evaluable patients (1 microsatellite unstable and 4 microsatellite stable).25 A trial combining immune checkpoint inhibition with interferon alpha-2b (ClinicalTrial.gov identifier: NCT02982720) is also ongoing.
Conclusion
Currently, unresectable cholangiocarcinomas are treated with cytotoxic chemotherapy, with variable responses. Recent genomic and immuno-profiling analyses have provided insight to the molecular heterogeneity of this tumor type as well as rationale for testing novel targeted and immunotherapeutic approaches in the treatment of subsets of cholangiocarcinoma. Specifically, IDH inhibitors, FGFR inhibitors, BRAF inhibitors, anti-HER2 directed therapies, and immune checkpoint inhibitors have demonstrated favorable safety profiles and clinical responses in patients with disease that is refractory or intolerant to chemotherapy. Results from randomized phase III studies will be helpful in determining how clinical outcomes with these targeted and immunotherapeutic agents compare to chemotherapy and will also help to better characterize the unique toxicity of these targeted agents. Biomarker identification is still needed to help identify the subset of patients who may benefit most from these novel approaches and rational combination strategies to help improve response rate and durability are currently underway. However, the recent data highlight the need to obtain molecular profiling on all newly diagnosed patients with unresectable cholangiocarcinoma to identify whether clinical trials with these novel agents should be considered.