Cancer immunotherapy has come of age and has successfully been implemented as the standard of care in a number of oncologic indications.1 Antibodies targeting cancer-associated antigens on the tumour cell, such as CD20, constituted the first wave of immunotherapies leading to the first approval of an antibody for cancer therapy. In 1997, rituximab, an anti-CD20 antibody was approved for the treatment of high-grade B cell lymphomas.2 Twenty-one compounds with similar tumour-targeted concepts have been approved for cancer treatment in Europe in the meantime. Despite being thought that the mode of action of such antibodies mainly relied on a direct antitumoural attack, emerging evidence suggests a contribution of the innate and adaptive immune system.3–6 Tumour-targeted monoclonal antibodies are now mainly considered as a passive immunotherapy.7–10
More recently, a paradigm change has occurred, moving the focus of therapeutic endeavours away from the cancer cell to effector components of the immune system, mainly T cells.11 Preclinical and clinical evidence have now demonstrated that allowing T cell activation results in antitumoural activity without bona fide tumour-targeting. Antibodies targeting checkpoint inhibitors on T cells such as, programmed death receptor 1 (PD-1) or cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), reverse T cell anergy and lead to T cell-mediated remissions. Such antibodies can induce unparalleled activity in patients with advanced stage disease or having failed multiple lines of therapy, even in entities classically deemed nonsuitable for immunotherapies. As a consequence, approvals for cancer immunotherapies now span a broad scope of indications including acute lymphatic leukaemia (ALL), melanoma, nonsmall cell lung cancer, kidney cancer, Hodgkin lymphoma and head and neck cancer.12–17 A major limitation of the approach is its unspecific nature of T cell targeting, resulting in severe side effects.18–20 Strategies allowing for a more directed and specific therapeutic use of T cells may have advantages in terms of specificity and efficacy.
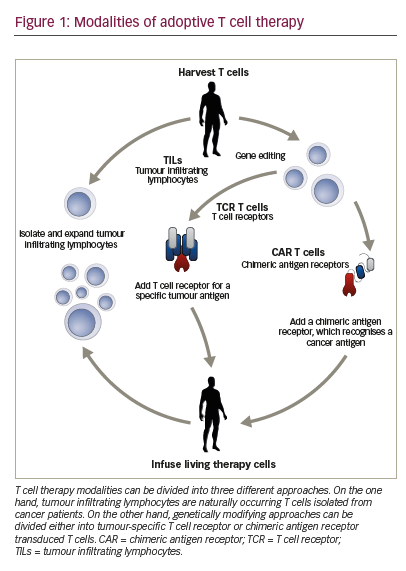
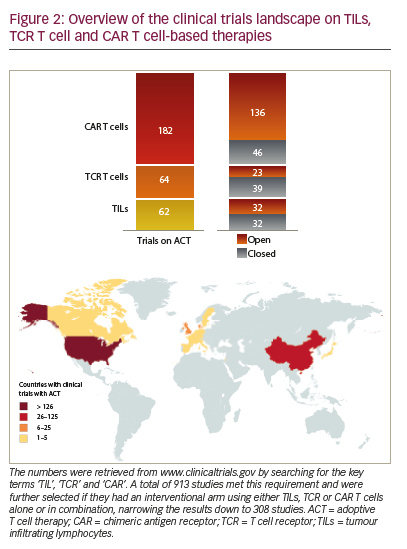
Adoptive T cell therapy (ACT) is a strategy that directly employs T cells with therapeutic intention against cancer.21 T cells can be isolated from a patient’s blood or tumour samples. Those derived from the tumour are also known as tumour-infiltrating lymphocytes (TILs).21,22 Isolation is followed by in vitro expansion and manipulation, and then reinfusion into the patient.21 ACT protocols that employ TILs are generally accompanied by preconditioning of the patients prior to treatment. In most cases, however, TILs cannot be isolated due to the lack of accessible tumour tissue or lack of tumour-specific T cells.23 Through genetic engineering, T cells can be rendered specific for a given target. This process encompasses the transduction or transient transfection with defined natural or synthetic genes. Two major strategies to engineer tumourspecific T cells have emerged: T cell receptors (TCRs)24–26 specific for a peptide presented in a major histocompatibility complex-dependent manner, and chimeric antigen receptors (CARs)27–30 which are synthetic T cell activating receptors targeting cell surface antigens. TCRs correspond to the natural molecule found in any T cell with modifications to enhance their biochemical and functional properties, while CARs are constituted of the variable fragment of an antibody fused to T cell activating CD3 zeta chain and co-stimulatory domains (overview in Figure 1). TILs, TCR and CAR T cells are currently being tested in various tumour indications in clinical trials (Figure 2).
Clinical efficacy of adoptive T cell therapy
ACT has so far been successfully applied in patients suffering from melanoma, myeloma, cervical cancer, lymphoma, bile duct cancer, neuroblastoma and others.21,31,32 For TIL-based therapies, highest clinical benefit has been observed in melanoma in conjunction with total body irradiation or nonmyeloablative chemotherapy as preconditioning regimens. In a study where 93 patients with metastatic melanoma
were treated with autologous TILs administered in conjunction with interleukin (IL)-2 following a lymphodepleting regimen, 20 patients achieved a complete and mostly lasting tumour regression. These 20 complete responders had 3- and 5-year survival rates of 100% and 93%.33 The antitumour effects of melanoma-derived TILs have encouraged trials in other indications with limited success. TILs have been extensively characterised through whole-exome sequencing and other approaches. A surprising feature was that few T cells of the TIL product were in fact tumour-specific or at least of definable specificity.34–36 The high response rates of patients with advanced melanoma to TIL therapy have prompted the initiation of a phase III clinical trial comparing TIL therapy to ipilimumab treatment as standard of care arm (NCT02278887).
TCR-modified T cells have so far mainly been used in phase I or II clinical trials. Early attempts using melanoma-associated antigen 3 (MAGE-A3)-specific T cells reported objective responses in five out of nine treated patients. However, at the same time, two patients died due to neurotoxicity, probably related to expression of a closely related antigen on the brain, highlighting the risks associated with high-affinity TCRs.37 Similarly, in another study two patients developed a cardiogenic shock and died within days of T cell therapy infusion with the autopsy showing myocardial T cell infiltration. No expression of MAGE-A3 on heart tissue was found but the TCR was then demonstrated to cross react with an epitope of another antigen, namely titin. This further stresses the potential difficulties of predicting off-target effects of such a potent therapy.38 Objective responses have also been reported using TCRs specific for MAGE-A4 in oesophageal cancer, for MART-1 in melanoma, for NY-ESO-1 in melanoma, myeloma and synovial sarcoma, as well as for carcinoembryonic antigen (CEA) in colon cancer.39–43 The most prominent responses have so far been seen in patients with melanoma
treated with NY-ESO-1-TCR-transduced T cells. The treatment regimen included IL-2 and preparative chemotherapy.40
CAR-transduced T cells have been mainly tested in patients suffering from haematologic malignancies. The most used target is CD19, a B cell lineage antigen, expressed in a wide variety of entities such as B cell lymphomas and B cell leukaemias. Several clinical CD19-CAR candidates developed by different groups are currently under investigation with similar but not identical structures nor sequences. In adult and paediatric ALL, CD19-CAR T cells have induced major clinical responses with complete remission rates as high as 90% (27 out of 30 patients).44 Similar results could be confirmed in independent trials using distinct CD19-CAR constructs.45 Data from other B cell malignancies could confirm CAR T cell activity but seem to indicate a trend towards enhanced response rates in more aggressive haematologic malignancies. For example, response rates for patients with diffuse large B cell lymphoma were four complete remissions out of seven patients treated (57%), as opposed to four out of 14 patients (29%) with chronic lymphatic leukaemia in another study.46 In contrast, efficacy of CAR targeting nonhaematologic malignancies and using different targets have proven significantly less successful with so far only little objective responses observed in clinical trials. CAR therapy has proven to have a manageable safety profile in these small cohorts and in specialised centres. However, several neurotoxicityassociated deaths have recently occurred in a single ongoing CD19- CAR T cell trial with a defined CAR which were not observed in other parallel trials (NCT01044069, NCT01840566, NCT02535364).47–49 Earlier, an ERBB2-recognising CAR induced a severe cytokine storm syndrome that led to death of one patient within 5 days of therapy. The authors postulated that the severe cytokine storm was induced by on-target offtumour recognition upon first-pass clearance in the lung. This cytokine storm caused pulmonary toxicity and oedema, resulting in multiorgan failure.50 While the first approval of a CAR therapy seems to be in sight, the toxicity together with the limited efficacy in solid tumours call for a better understanding of the underlying mechanisms of action as well as strategies to overcome their inherent limitations.
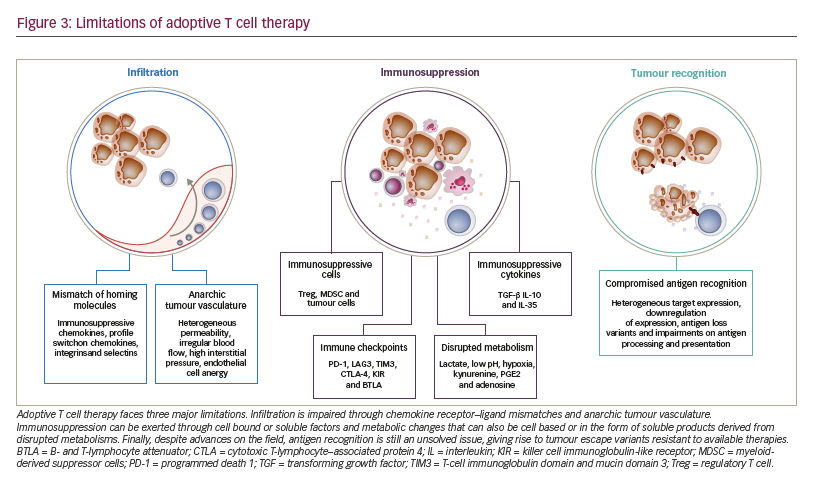
Limitations to adoptive T cell therapy efficacy Safety of application is the major limitation to ACT and has been extensively reviewed recently.51 After the T cells have been administered to the patient, three major mechanisms limit their antitumoural potency and efficacy (Figure 3):
• limited access to the tumour tissue;
• limited recognition and lysis of tumour cells; and
• local immune suppression.
The process of distribution of T cells to the tumour site and subsequent activity is complex, requiring migration, persistence, and activation of the T cell in a given tissue.28 As T cells are natural antitumoural effectors, strongly involved in the editing and elimination phases of cancer development, tumours have mounted complex strategies to selectively exclude T cells from their vicinity.52 Mechanisms include pathologic vascularisation limiting T cell influx and extravasation,53 selective expression or repression of integrins, or production of chemokines excluding T cell attraction.54–58 Along these lines, even T cells that have managed to enter tumour tissue are limited in their activity due to a lack of tumour recognition. The latter being due to heterogeneous target expression, downregulation of expression, antigen loss variants (as a result of either self-mimicry or tolerance) or impairments on antigen processing and presentation.59,60 On the other hand, even in the case of effective tumour recognition, tumour cells are still able to repress T cell activation. Tumours can suppress T cell function through soluble or cell bound factors, as well as the induction and attraction of suppressive cell populations. This local immune suppression, which may also escalate to become systemic, includes factors such as transforming growth factor beta (TGF-β), indoleamine 2,3-dioxygenase (IDO), PD-1-PD-L1 interactions, myeloidderived suppressor cells or regulatory T cells.6,56,61
A major focus of development over the past years has been on the enhancement of tumour recognition through discovery of new target epitopes for CARs62 (listed on the Cancer Immunity Peptide Database63) and additional engineering of these receptors with series of modifications on co-stimulatory molecules,64,65 hinge regions66 and transmembrane regions.67–73 However, the clinical value of these modifications is unclear, as comparative clinical studies have yet to be pursued. As T cells used in ACT are, in general, as sensitive to suppression as endogenous cells, a growing body of evidence points towards the combination of ACT with immune-activating strategies. Preclinical evidence indicates that combination of ACT with immune checkpoint blockade will overcome immune suppression and lead to synergistic activity.74 Several clinical trials have been launched to test the combination of TCR, CAR or TIL ACT with checkpoint blockade (NCT02652455, NCT03030001, NCT02873390, NCT02862028, NCT02858310, NCT02775292, NCT02070406). Solid tumours have been recognised as entities that do not respond significantly to CAR ACT. Comprehensive overviews of clinical ACT experience in solid tumours have been recently published. Johnson and June have reviewed current ACT clinical trials on solid tumours and compared TCR to CAR T cell trials,75 and Yong and colleagues have circled the limitations of ACT in solid tumours.76
While the main efforts in the past years to improve ACT have been put on tumour recognition and immune suppression, less emphasis has been put on the prerequisite for either mode of action: the access of T cells to the tumour tissue. Here we will summarise available evidence to tackle this central issue.
T cell migration and entry into tumours
In cancer immunotherapy, T cell trafficking has recently come into spotlight due to the findings that higher TILs correlate with better clinical outcomes.77 This is particularly evident for gliomas,78 colorectal,79 breast80 and cervical cancers.81 An international consortium is currently prospectively validating this prognostic parameter as an ‘immunoscore’ for colorectal cancer. Such a score may, in the near future, be used in clinical routine.82 This body of evidence has confirmed the preclinical observations that high T cell infiltrations are required for adequate antitumoural activity of T cells. To gain access to tumours or to any tissue, T cells need to pass through the two-step process of leukocyte– endothelial cell interaction proposed by von Andrian and colleagues.83,84 This model postulates that selectins play a crucial role on the first adhesion of leukocytes to the endothelial walls. In a rather weak manner, this selectin binding allows cells to slow down and form more stable bridges though second mediators, integrins, that will subsequently be responsible for the trafficking of leukocytes.83 The multistep adhesion cascade, comprises three key steps of rolling, adhesion, and migration requiring G protein-coupled receptors for the activation of the integrin signal on T cells.85–89 These particular G protein-coupled receptors are also referred to as chemokine receptors and control migration.90–94 Chemokine receptors include four conserved cysteine residues in their sequences, which form different patterns of disulphide bonds. The spacing of two relevant cysteines defines five families: the CC-, CXC-, XC-, CX3C- and CX- family of chemokines. An L is added to identify ligands, and within families different proteins are distinguished with numbers.95,96
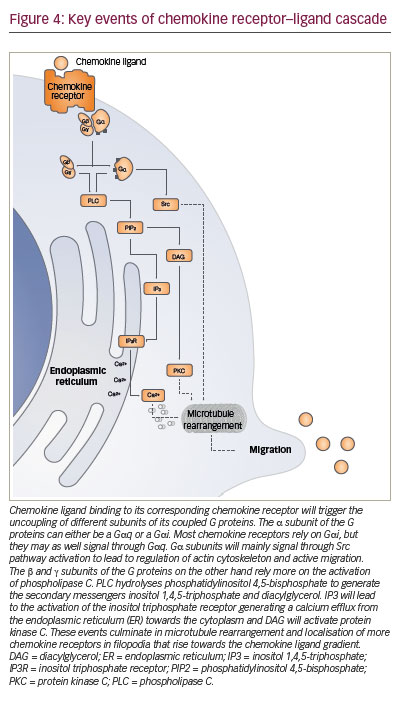
The key signalling event triggered intracellularly after the binding of chemokine ligand to its cognate receptor is a cytosolic calcium elevation (Figure 4). The change of conformation in the chemokine receptor allows activation of the G protein that, in turn, will activate phospholipase C. This cleaves phosphatidylinositol diphosphate into diacylglycerol, that activates protein kinase C, and into inositol triphosphate which triggers calcium elevation. This chain reaction wind up in signalling via MAPK, ERK and JAK, which induce the expression of other proteins promoting cell migration.97–99
Tumours shape their chemokine milieu by excluding T cells, recruiting suppressive cell populations, triggering metastasis and disrupting vasculature anatomy
Cells of different tumour entities will express different patterns of chemokines such as CCL2 to CCL5, CCL8, CCL9, CCL17, CCL19, CCL21, CCL22, CCL26 to CCL28, CXCL1 to CXCL8 and CXCL12 to shape their tumour microenvironment.100,101 By altering the levels of key chemokines, some tumours are able to avert immunologic surveillance of tissue resident lymphocytes. For example, certain types of ovarian and breast cancers have been proven to upregulate CCL22 and CCL28,102,103 which are central chemokines for the recruitment of regulatory T cells.104 In colorectal tumours there is upregulation of CCL2, a chemokine central for the recruitment of tumour-associated macrophages and T cells.105 Melanomas show an even broader response by decreasing expression patterns of CCL2, CCL3, CCL4, CCL5, CXCL9 and CXCL10 55,106 which control the recruitment of not only T cells, but also of proinflammatory immune cells, thereby contributing to immune evasion. As further evidence that the modulation of chemokine patterns might be used by tumours to control the trafficking of T cells the induction of specific chemokine ligands in melanoma triggers the recruitment of T cells to the tumour site.55 Tumour-derived microvesicles have been shown to induce secretion of CXCL8, CCL2, CCL3, CCL4 and CCL5.107
![]()
CCL2, CCL5, CXCL5 and CXCL12, have been shown to promote tumour progression by increasing the formation, recruitment and suppressive activity of tumour-associated macrophages and myeloid-derived suppressor cells.108–110
Besides shaping their environment to reduce the number of infiltrating effector T cells, tumours may also take advantage of chemokine receptors to initiate metastasis. CCL5 has been found to be produced in prostate cancers and osteosarcomas improving the survival of tumour cells through increased angiogenesis.111 Myeloid cells that produce metalloproteinases 2 and 9 have been found to migrate towards CCL9 in colon cancer models potentially improving invasive growth of tumours.112 CXCR4 expression on tumour cells, on the other hand, is associated with metastasis from prostate cancer, lung cancer and glioblastoma even though this receptor is not expressed physiologically in all of these tissues.113 The axis CCL1– CCR8 is believed to play a role in initial stages of metastasis from the primary tumour site to the subcapsular sinus of lymph nodes.114 Several of the chemokines previously mentioned also have angiogenic properties: pro-angiogenic properties have been ascribed to CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, CXCL8 and CXCL12, whereas anti-angiogenic properties have been associated to CXCL9, CXCL10, CXCL11 and CXCL14.115 Disruption of these pathways will result in defective or anarchic tumour vasculature that will hamper migration of the immune effector cell to the tumour site.116,117 In summary, available data strongly suggests that chemokines are key players in cancer progression by several mechanisms. As a consequence, therapeutic strategies modulating this axis may restore immune surveillance and T cell efficacy.
Modulation of the local chemokine milieu to recruit T cells to the tumour
Therapeutic approaches have shown the potential of modulating the tumoural chemokine milieu to either improve the trafficking of effector cells or to decrease the trafficking of immunosuppressive cells towards the tumour. Small molecules blocking CCR4 decrease the amount of T regulatory cells in tumours, antagonising CCL17- or CCL22-mediated Treg-recruitment to the tumour.118–120 In vitro, some truncated chemokines can act as antagonists to their cognate receptors such as CXCL8, CCL5 and CCL2.121–123 Several antibodies targeting chemokine ligands are being tested for tumour immunotherapy. However, only a truncated analogue of CCL2 has shown efficiency in decreasing the recruitment of tumour-associated macrophages.124,125 An antibody targeting CCR4, mogamulizumab, is approved in Japan for the treatment of T cell lymphoma.126 Other antibodies targeting different chemokines and chemokine receptors for tumour immunotherapy have been developed and are currently in clinical trials.125
Chemokines and chemokine receptors in the tumour tissue may also be induced in the course of therapy: treatment of tumour-bearing mice with chemotherapy (dacarbazine, temozolomide and cisplatin) induced intratumoural expression of CXCR3 and CCL5, enabling T cell infiltration into cutaneous tumours.106 PD-1 pathway blockade has been reported to increase IFN-γ at the tumour site, thereby increasing chemokinedependent trafficking of immune cells into malignant disease sites.127 Heparins can inhibit interferon gamma (IFN-γ)-induced chemokines, namely CXCL9 and CXCL10 and lead to recruitment of pro-inflammatory immune cells.128 CCL1 blockade has proved in murine models to reduce intratumoural accumulation of T regulatory cells decreasing the immunosuppression in the tumour and allowing more effective T cell function.129 In ovarian cancer models, the infusion of NKG2D-CARexpressing T cells increased the number of host CD4+ and CD8+ T cells at the tumour site in a CXCR3-dependent manner and increased the number of antigen-specific host CD4+ T cells in the tumour and draining lymph nodes. This showed that adoptively transferred T cells recruit and activate endogenous T cells. This contributes to the elimination of tumour cells and to the development of tumour-specific memory responses.130 Other forms of immunotherapy also have the capacity of changing the chemokine receptor–ligand pattern in tumours such as Listeria monocytogenes-based anticancer vaccine131 or even systemic IL-12 treatment.132 It is surprising that, to date, no clinical trial has yet been initiated to investigate therapeutic interference with the chemokine pattern in combination with ACT, aside from – indirectly – when administering checkpoint blockade.
T cell engineering with chemokine receptors for enhanced tumour-directed migration
Upon development in primary lymphoid structures, T cells only obtain a limited amount of information about self and nonself antigens. They have not completed their specificity maturation until they encounter antigen-presenting cells at secondary lymphoid structures.88 This step is known as priming133 and it allows T cells to tune their activity against a specific antigen and to expand their cytolytic potential.134–137 Another function of the encounter of the antigen-presenting cell with the T cell is to regulate their subsequent homing through molecular migration signatures for selected tissues.138–143 As a consequence, T cells will only express a limited repertoire of chemokine receptors naturally. Therapeutic transduction of T cells with defined chemokine receptors and thus modulation of their migration signatures may enhance efficacy of ACT.
Kershaw and colleagues were the first in 2002 to arm T cells with chemokine receptors to make use of CXCL1 overexpression in tumours by tumour cells. As the corresponding receptor, CXCR2, was not found on T cells, the authors retrovirally transduced T cells to overexpress this receptor.144 In murine tumour models they demonstrated the feasibility of changing the chemokine receptor profile of T cells towards chemokines secreted by tumours. The value of CXCR2 as a chemokine receptor of choice was later confirmed by using an approach of mRNA transfection, potentially offering superior safety.145
The combination of CAR T cell therapy and chemokine receptors has repeatedly proven to be synergistic or at least additive in terms of activity in preclinical trials. Co-expression of CCR4 on CAR-CD30-modified T cells improved homing to CD30+ Hodgkin lymphoma, consequently improving the effectiveness of ACT.146 In a neuroblastoma model, expression of CCR2b redirected GD2-CAR-transduced T cells generating greater anti-tumour activity.147 High secretion of CCL2 by malignant pleural mesotheliomas allowed for the use of this chemokine gradient to recruit gene-modified T cells transduced with CCR2. These cells also carried a CAR construct that conferred them antigen specificity for mesothelin. CAR T cells that bore a functional chemokine receptor were able to overcome the inadequate tumour localisation that limits conventional CAR targeting strategies and can significantly improve antitumour efficacy in vivo.148 Our group has shown that CCR4 transduction enhances the migration of T cells to the tumour microenvironment in a pancreatic tumour model (Figure 5). The cytolytic activity of engineered T cells was increased, stressing the importance of considering not only tumour cells as targets, but also other immune infiltrating cells.104
Engineering through CX3CR1 improved tumour growth control with higher infiltration rates of CX3CR1-engineered T cells in a gradient-dependent manner in human colorectal cancer models in immunodeficient mice.149
Natural killer (NK) cells have also been of interest for migration modulation through chemokine receptors. CCR7 increased the efficacy of NK cells that had further been engineered with the high-affinity antibody-binding receptor CD16, against rituximab-coated lymphoma cells.150 Xu and colleagues have engineered a photoactivatable-CXCR4 that reacts by directing the migration of cell towards 505 nm light.151
Despite the success so far, there is a limited number of current clinical trials with chemokine receptor-transduced T cells. The MD Anderson Cancer Center is currently running a clinical trial with TILs transduced with CXCR2 (NCT01740557). The goal of this clinical trial is to assess the side effects of CXCR2-transduced TILs in combination with chemotherapy (cyclophosphamide and fludarabine) and aldesleukin, in patients with metastatic melanoma, in an attempt to allow them to better localise the tumour. The study has the toxicity of CXCR2-transduced TILs as its current primary outcome and tumour response using Immune-Related Response Criteria as a secondary outcome. Only one control arm has been set for this study, with each patient receiving a therapy consisting of both CXCR2-transduced TILs and nerve growth factor receptor (NGFR)- transduced TILs (control cells).
Modulation of T cell trafficking beyond chemokine – chemokine receptor interactions
The possibilities for controlling T cell trafficking and redirecting them to the tumour are broader than the direct or indirect modulation of chemokine ligands and receptors (Figure 6). Different chemotherapies given in low doses also have the potential, besides the previously mentioned capacity of modulating the chemokine milieu, to selectively decrease immunosuppressive cells and induce an anti-angiogenic activity.152 The combination of doxorubicin and IL-12 improved the trafficking of NKG2D T cells towards the breast tumours.153
Delivery of IL-12 into murine tumours using T cells genetically engineered to express an anti-vascular endothelial growth factor receptor (VEGFR)-2 CAR lead to tumour regressions without the need for IL-2 regimens. Anti-VEGFR-2 CAR and IL-12-co-transduced T cells infiltrated tumours, expanded, and persisted for prolonged periods.154 VEGF expression in tumours correlates with poor levels of infiltrating T cells,155 and, vice versa, bevacizumab, an anti-angiogenic antibody improved T cell infiltration into solid tumours.156 Other antiangiogenic drugs, namely anginex, endostatin and angiostatin enhanced T cell infiltration in murine colon cancer and melanoma models.155,157
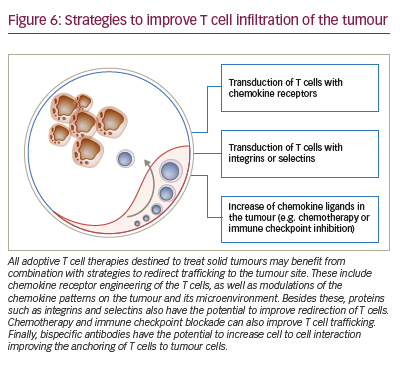
Radiation also alters tumour vasculature and can improve the access of T cells to tumours.158 On another line, the disruption of tumour function of endothelin B receptor can facilitate the access of T cells through the endothelial barrier.159 Improving homing through integrins has proved successful in preclinical models with T cells engineered with integrin alpha v beta 3 showing benefits in vascularised solid tumours.160 Immune checkpoint blockade, besides directly allowing the intratumour activation and expansion of T cells, also increases production of IFN-γ-induced cytokines and chemokines. This leads to the recruitment of more effector cells to the tumour site.161 Our own group showed, that in a murine gastric cancer model, a bispecific antibody targeting on the one hand EGFRtransduced T cells, and on the other hand EpCAM bearing tumour cells increases recruitment of these engineered T cells to the tumour site.162 Bispecific antibodies targeting tumour cell and T cell simultaneously are tested in clinical trials on patients whose tumours express different antigens such as EpCAM, HER2, CEA and PSMA (NCT00635596, NCT01284231, NCT01723475, NCT00351858).
Finally, in rare cases, the issue of migration into the tumour after systemic administration of T cells may be overcome through local administration of CAR T cells.163 This results in higher accumulation at tumour sites and superior control of tumour growth.164 This strategy is, however, only envisionable when the tumour site is readily accessible. This is the case, for example, for malignant pleural mesothelioma where the pleural space is directly accessible through a minimally invasive puncture.
Conclusion and outlook
TIL, TCR-transduced T cell and CAR-transduced T cell therapies are under clinical investigation for cancer treatment. Current clinical trials will decide on their approval for certain indications. While some of these strategies are likely to be approved in defined indications such as ALL, the majority of tumour patients will not benefit from this approach in the near future. A major reason, especially for solid tumours, is the limited access of adoptively administered T cells to the tumour tissue. Strategies such as T cell engineering with chemokine receptors need to be tested in clinical protocols based on preclinical evidence. However, it needs to be stressed, that those cells that eventually reach the tumour, still need to recognise the tumour cell adequately and prevail over immunosuppression. This will require both advanced T cell engineering and combination with other immune and nonimmune therapies.