Advanced non-small cell lung carcinoma (NSCLC) treatment paradigms have evolved during the past decade. Identification of tumor-specific molecular alteration in cancer driver genes has led to the development of targeted therapies.1–3 Most of the tumors harboring such alterations are sensitive to tyrosine kinase inhibitor (TKI) drugs, making such oncogenic drivers promising targets for the development of antitumor therapeutics.4,5
MET is a proto-oncogene that can act as an oncogenic driver after certain genomic alterations. It is expressed in many epithelial as well as mesenchymal cells, including hepatocytes, hematopoietic cells, and neuronal cells, and is essential for important biological processes, such as embryonic development and organogenesis.6,7 However, mutations and its aberrant activation can promote tumor development and cancer progression by dysregulating downstream signaling pathways.8,9 Initially, abnormal MET signaling was believed to be the mechanism of resistance acquired by NSCLC tumor cells against certain therapeutics.10–12 Further reports demonstrated the role of MET alterations in sustained MET pathway dysregulation, leading to oncogenesis.13–15 Clinically, NSCLCs with MET alterations are associated with poor prognosis, and these alterations have been recognized as an important therapeutic target in various cancers, including NSCLC.16–18
In this review, we discuss the current understanding of the implications of aberrant MET activation in NSCLC harboring MET exon 14 (METex14) skipping alteration, available diagnostic options, potential therapies in the pipeline, and the future clinical landscape.
Structure and function of the MET receptor
MET was first identified in a chemically treated human-osteosarcoma-derived cell line as a transforming gene from a fusion of TPR-MET.19 The MET gene is located on chromosome 7q31 in the human genome, which spans about 125 kb DNA and contains 21 exons and 20 introns.20 MET is encoded as a precursor, which is modified into a mature protein by proteolytic cleavage between its a and b subunits.21 A mature MET protein is composed of a small a subunit (50 kDa) and a larger b (145 kDa) subunit linked together by a disulfide bridge.8 The a subunit and a portion of b subunit together form the extracellular region of the heterodimer protein, while the remainder of the b subunit comprise the transmembrane and intracellular regions (Figure 1A).
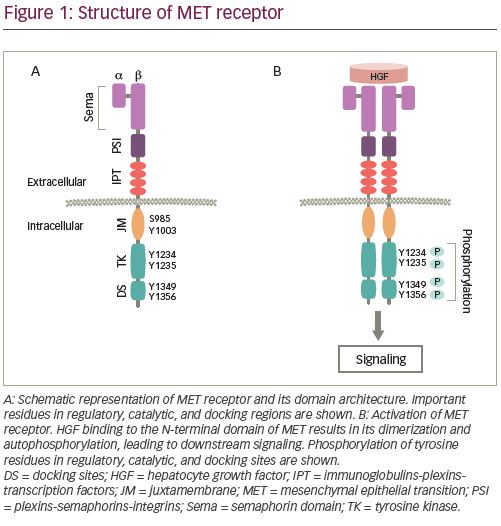
The extracellular component of MET contains three domains. N-terminal Sema (Sema-phorin) is the largest domain comprising 500 residues, which encompasses the a and a part of b subunits. The domain is essential for the ligand binding,22 dimerization, and activation of MET.23,24 The Sema domain is followed by the plexins-semaphorins-integrins (PSI) domain, containing four disulphide bonds, which are essential for the proper orientation of the receptor for ligand binding.25 The PSI domain is connected to the transmembrane helix of MET through the immunoglobulin-plexins-transcription factor domain. The intracellular portion of the receptor includes a juxtamembrane (JM) domain, a tyrosine kinase (TK) catalytic domain, and a C-terminal multifunctional docking site.22 Binding of its ligand, hepatocyte growth factor (HGF), which is also known as scatter factor, is essential for the activation of the kinase activity.26,27 HGF is the only MET receptor ligand known so far and binds to the receptor with high affinity.22,28
MET signaling and its dysregulation in NSCLC
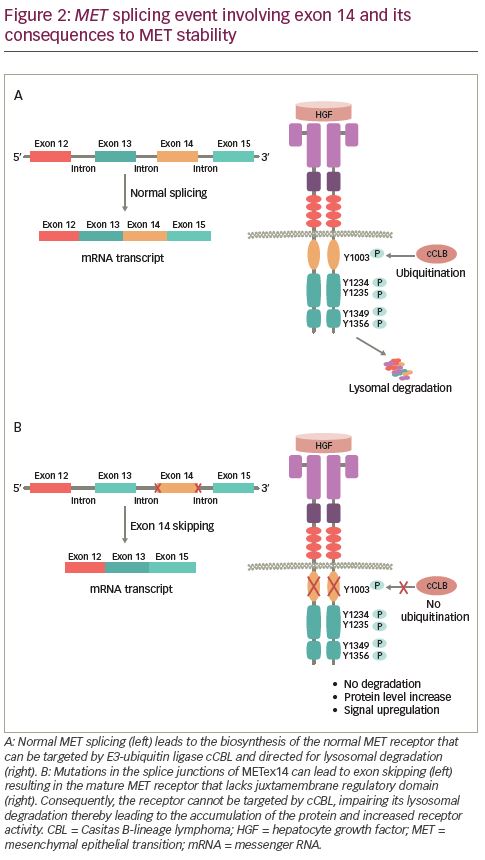
HGF binding to MET causes dimerization of the receptor leading to the autophosphorylation of intracellular residues Y1234 and Y1235 in the kinase domain followed by phosphorylation of two additional tyrosine residues, Y1349 and Y1356, in the C-terminal outside of the kinase domain (Figure 1B). Phosphorylation of the C-terminal residues leads to the formation of the docking site, which is necessary for the engagement of signaling partners.29 Subsequently, adapter and effector proteins, such as GRB2 (growth factor receptor bound protein 2), GAB1 (GRB2 associated binding protein 1) and SHC (Src homology 2 domain-containing), bind to the docking site triggering downstream signaling.30–36 MET signaling plays a crucial role in executing various cellular functions.37–39 To maintain functional balance and cellular integrity, MET activity is regulated through various mechanisms. The active MET receptor can phosphorylate at residue Y1003 in the JM domain, a site for the recruitment of E3-ligase Casitas B-lineage lymphoma (CBL), and subsequently undergo ubiquitin-mediated lysosomal degradation, leading to the downregulation of MET (Figure 2A).40–42 Additionally, it has been shown that phosphorylation of S985 at JM domain acts as a counterbalance to receptor activation, by negatively regulating its activity, even in the presence of HGF.43,44 Furthermore, proteolytic cleavage of MET by ADAMs (a disintegrin and metalloproteinase) and gamma-secretase may also contribute to the downregulation of MET receptor activity.45,46
Alterations in MET can result in the dysregulation of MET signaling, which is present in various solid tumors including NSCLC and is associated with tumor progression and metastasis.47–49 Gene amplification, rearrangement, and skipping alterations, which lead to the overexpression and impaired degradation of MET, are the major underlying factors of aberrant MET activation.50,51 Alteration or deletion of crucial residues in regulatory domain interfere with mechanisms that help to maintain MET receptor turnover leading to its accumulation and hyperactivation.52–54
METex14 skipping alteration in NSCLC
Skipping of METex14 in NSCLC was first reported in 2005.55 Substitutions or deletions at 3′ splice site in intron 13 or the 5′ end splice site of intron 14 results in METex14 skipping.56,57 This somatic alteration, at or around the splice junction of METex14, leads to the loss of exon 14 in the transcript and synthesis of the MET protein with an in-frame deletion of 47 amino acids in the JM domain (including residue Y1003) ablating the CBL-mediating ubiquitination and degradation of the receptor (Figure 2B).24,58 Consequently, METex14 skipping results in increased levels of MET protein, which can drive activation of downstream signaling pathways that promote tumor development.57,59
Splicing occurs through two sequential steps involving various parts of the intron. The splice donor site and the splice acceptor site are present at the 5′ and 3′ ends, respectively. The splice acceptor site is flanked upstream by the branch point and poly-pyrimidine tract sites (Figure 3A). First, the branch point nucleotide performs a nucleophilic attack on the first nucleotide of the intron at the splice donor site. This forms an intermediate loop or lariat. Subsequently, the 3′ end of the released exon performs a similar nucleophilic attack on the last nucleotide of the SA thereby fusing the exons and releasing the intron lariat.60,61 Most METex14 skipping alterations involve the branch point, poly-pyrimidine tract or splice acceptor site in intron 13 or the splice donor site in intron 14. As shown in Figure 3B, these alterations interfere with the splicing mechanism leading to exon 14 skipping.
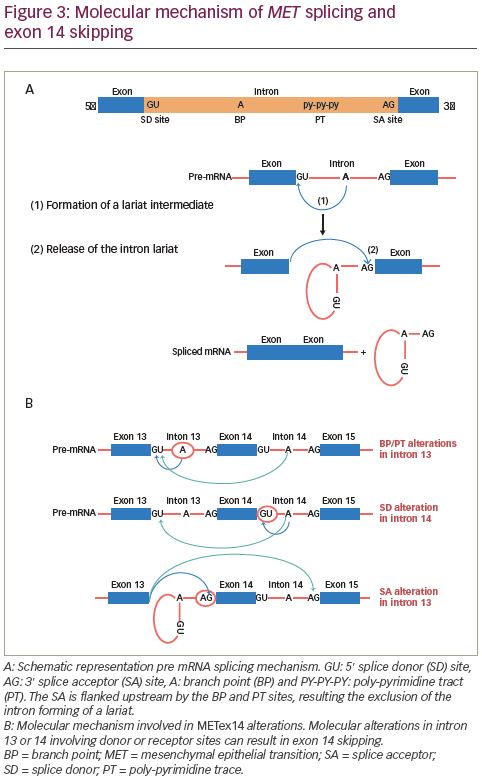
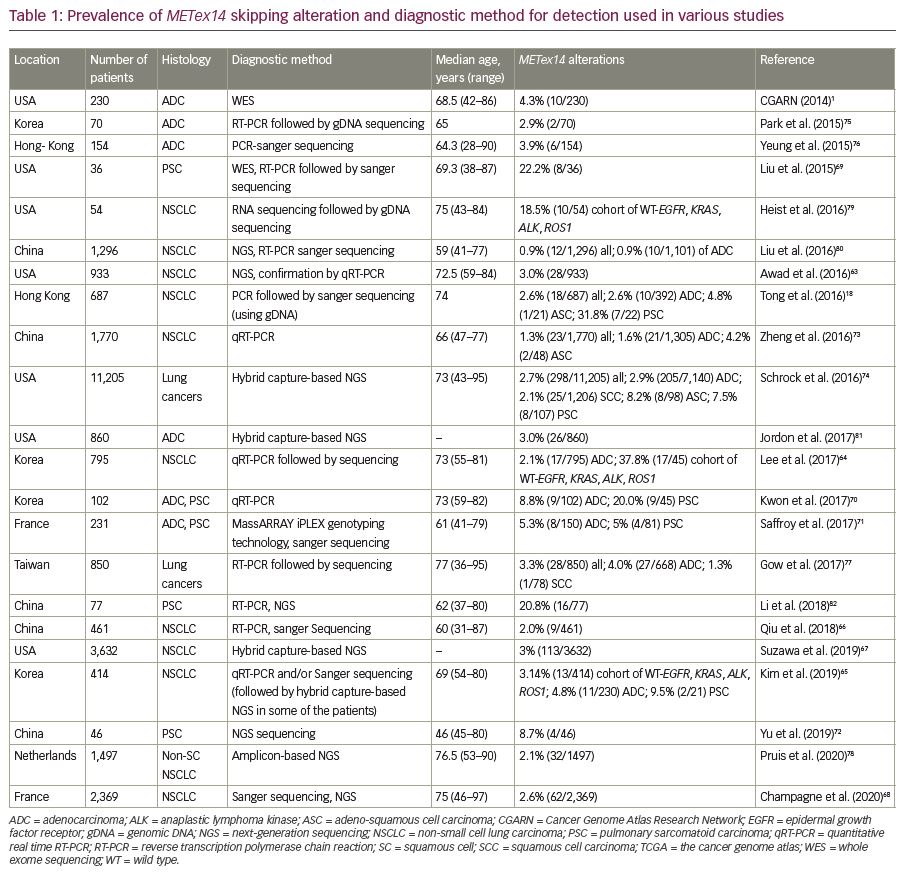
Interestingly, METex14 skipping alterations are primary oncogenic drivers in NSCLC, as these alterations are most likely to be mutually exclusive to other known oncogenic drivers, such as KRAS, EGFR, ALK, ROS1 or RET.57,62,63 Approximately 3–4% of NSCLCs harbor the METex14 alteration (Table 1).18,57,63–68 They are associated with some histologic subtypes of NSCLC but are not related to tumor stage. Among the histological subtypes, METex14 skipping alteration is commonly found in sarcomatoid carcinoma (4.9–31%),69–72 adenosquamous carcinoma (4–8%),18,73,74 adenocarcinoma (3–4%),1,18,57,65,75,76 and squamous cell carcinoma (2%).74,77 Also, among adenocarcinomas, the predominant subtypes are acinar (35–52.9%) or solid subtypes (35.3–53%).64,70,73,74,77 Clinically, METex14 skipping abnormality is found mostly in patients of advanced age.18,63,68,70,73,74,77,78
Detection of METex14 skipping alteration
Immunohistochemical analysis is a routine practice for the detection of MET overexpression. However, this technique on its own cannot specifically confirm METex14 skipping or an underlying alteration. Therefore, DNA- and RNA-based molecular assays are preferred methods for the detection of METex14 alteration. DNA-based sequencing assay can detect MET alterations such as insertions, deletions, point mutations, or duplications in splice sites, which may cause exon 14 skipping. Identification of such mutational hotspots leading to METex14 skipping alteration are used to predict the possible skipping event. However, METex14 skipping is associated with more than 120 reported sequence variants in splice sites, which makes it challenging to detect these mutations using only DNA-based assays.57,63 Therefore, analysis of RNA transcripts allows for the verification of fusion between exons 13 and 15.83 In ideal cases, both DNA- and RNA-based assays are used to complement each other for reliable detection of METex14 alterations (Table 1).1,18,63–82
Reverse transcription polymerase chain reaction (RT-PCR), quantitative real time RT-PCR, and Sanger sequencing are the routine approaches used for the analysis of mutations and METex14 alteration.18,73 mRNA transcript can be reverse transcribed using RT-PCR and corresponding complementary DNA is sequenced using Sanger techniques to verify exon 14 skipping from the sample. However, efficiency of the method relies on the quality of RNA, which is often derived from formalin-fixed paraffin-embedded or frozen tissue.83 Sanger sequencing of METex14 and its splice sites is still in routine practice for small scale analysis covering a portion of genomic region, which is performed using the PCR amplicon from genomic DNA covering exon 13 and exon 15, or cDNA from MET transcript. However, the European Society for Medical Oncology (ESMO) guidelines has proposed next-generation sequencing (NGS) and RNA sequencing, if possible, to detect METex14 alteration in its updated guidelines on September 2020.84,85
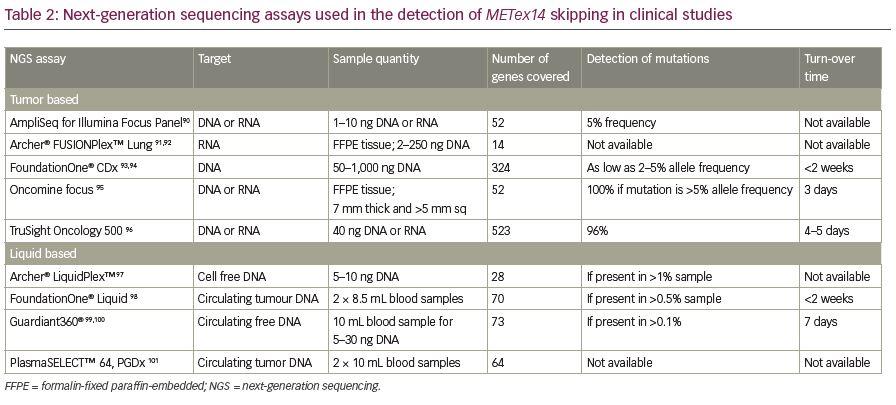
Recently, NGS has become a common diagnostic method to identify METex14 alterations. This high throughput method allows the large-scale analysis of multiple samples in a short time with comprehensive genomic coverage.1,64,86,87 The two most popular NGS sequencing panels used for targeted sequence profiling are hybridization capture and amplicon-based sequencing panels. The hybridization capture panel allows more comprehensive profiling for all alteration types, whereas the amplicon-based panel is ideal for analysing single nucleotide variants and indels (insertions and deletions). NGS analysis also can simultaneously detect other mutations or translocations (such as ALK, ROS1, RET, NTRK1, and NRG1 fusions) in a single assay.83 Due to the inherent difficulties in acquiring sufficient RNA material for testing, DNA-based NGS panels are used more frequently to identify METex14 skipping alterations. Recently, the US Food and Drug Administration (FDA) approved FoundationOne® CDx (Foundation Medicine, Cambridge, MA, USA) as a companion diagnostic test for this indication.88 Circulating tumor DNA (CtDNA) or RNA from plasma/blood samples (liquid biopsy) can also be used to identify METex14 alterations using NGS technologies. A clinical trial (VISION; ClinicalTrials.gov Identifier: NCTO2864992) aiming to test METex14 skipping alterations in circulating free DNA using plasma liquid biopsy is ongoing.89 Some of the commercially available targeted NGS assays that are used to detect these alterations are compared in Table 2.90–101
Therapeutic intervention of NSCLC with METex14 skipping alteration
NSCLC characterized with METex14 skipping alterations is targetable.57 Although many METex14 skipping tumors were found to express programmed death-ligand-1 (PD-L1), the overall response rate to PD-1/PD-L1-directed immune checkpoint inhibitors has been found to be low, and median progression-free survival (mPFS) was found to be short in patients with NSCLC.102,103 It should be noted that the mutation burden is generally low in such tumors. There are three therapeutic approaches to target tumors harboring METex14 skipping alteration:
- anti-MET and anti-HGF antibodies targeting the extracellular domain of the receptor;
- MET TKIs targeting the intracellular ATP binding pocket of target kinase to inhibit the autophosphorylation of the receptor; and
- antibody–drug conjugates.65,104,105
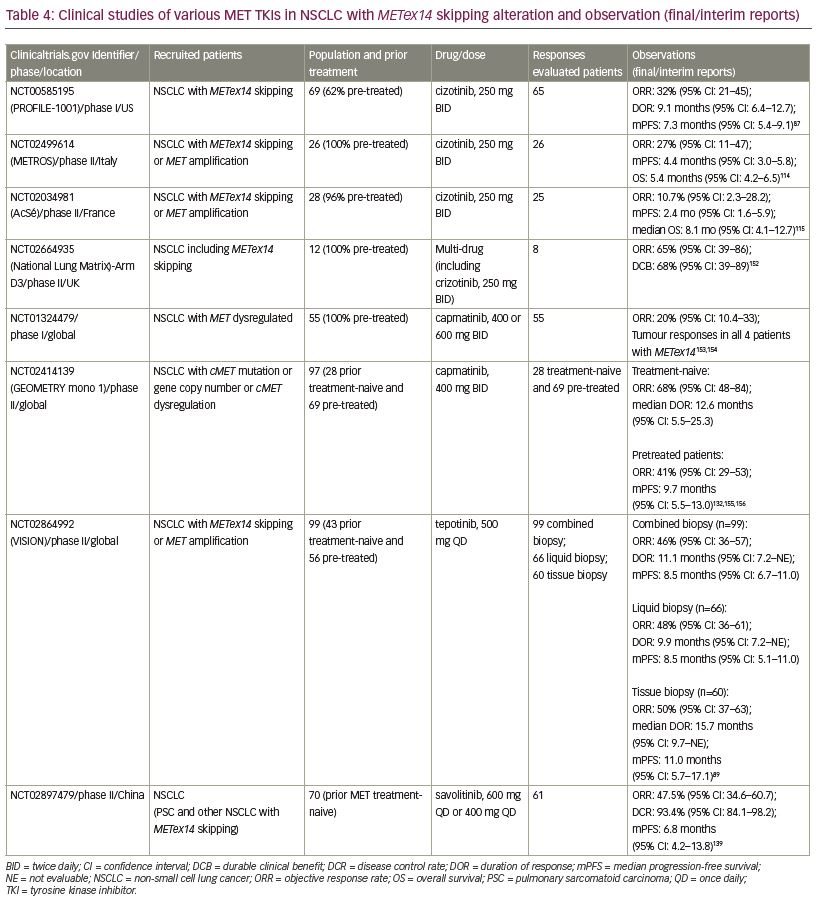
Clinical trials and case-reports have suggested varying degrees of responsiveness to experimental and FDA-approved small molecule TKIs against METex14 skipping NSCLC (Table 3). A multicenter retrospective analysis determined that the treatment with a MET TKI was associated with a significant prolongation in survival with a hazard ratio of 0.11 compared to patients who did not receive any MET inhibitor.106 MET TKIs are commonly divided into two types based on their targeting mechanism. Type I MET TKIs—such as crizotinib, capmatinib, tepotinib, and savolitinib—bind to MET in its catalytically active conformation where the aspartic acid-phenylalanine-glycine (DFG) motif projects into the ATP-binding site (DFG-in).87,107,108 Type II MET TKIs—such as cabozantinib, merestinib, and glesatinib—bind to MET in its inactive DFG-out conformation.109,110 Type I MET TKIs are further subdivided into type Ia (crizotinib) and Ib (capmatinib, tepotinib, and savolitinib) based on the interaction of TKI with G1163, a solvent residue. Various MET TKIs currently being used in clinical trials are listed in Table 4.
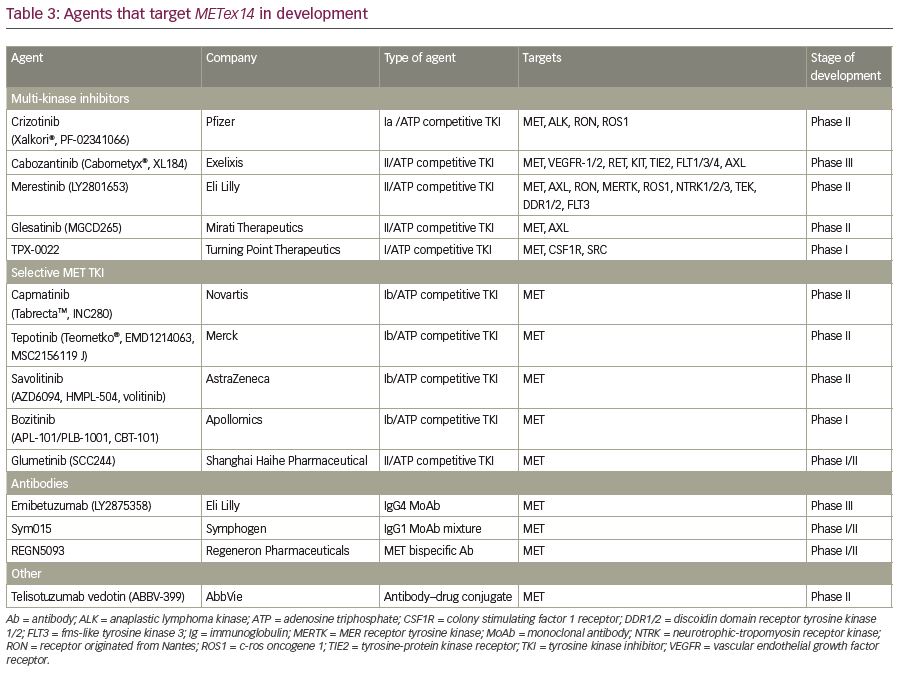
Multi-kinase MET inhibitors and their response against METex14 skipping NSCLC
This group of inhibitors targets multiple TKs and has been used to target MET kinase. Crizotinib, cabozantinib, merestinib, glesatinib, and TPX-0022 are the major target agents in this group.
Crizotinib
Crizotinib (PF-02341066; Xalkori®, Pfizer, New York, NY, USA) was originally developed as a MET inhibitor, which showed activity against ALK and ROS1 rearrangement, and was approved as an ALK and ROS inhibitor in NSCLC.63,64,111,112 Crizotinib was reported to have potent antitumor activity in NSCLC harboring MET amplification and exon 14 skipping alteration.57,87,113 The phase I study PROFILE 1001 (ClinicalTrials.gov Identifier: NCT00585195) was expanded to evaluate the efficacy and safety of crizotinib in 69 patients with NSCLC with METex14 alteration. Among 65 response-evaluable patients, 5% had a confirmed complete response, 28% had a confirmed partial response, 45% had stable disease, and mPFS was 7.4 months.87 In 2018, crizotinib received FDA breakthrough therapy designation for the treatment of patients with NSCLC with METex14 alterations based on a promising response rate of up to 44% in an earlier-phase study.59 A retrospective study of 22 patients treated with crizotinib reported a similar mPFS of 7.4 months.106 However, a phase II study (METROS; ClinicalTrials.gov Identifier: NCT0249961) reported an objective response rate (ORR) of 27% with an mPFS of 4.4 months in patients with NSCLC (n=26) with MET dysregulation with crizotinib.114 Similarly, the AcSé phase II study (by French National Cancer Institute; ClinicalTrials.gov Identifier: NCT02034981) reported insufficient ORR (10.7%), even after two cycles of crizotinib in 28 patients with NSCLC with METex14 alteration.115 Furthermore, neoadjuvant treatment with crizotinib in a locally advanced unresectable METex14 mutated lung adenocarcinoma converted the unresectable tumor into a resectable one.116
Cabozantinib
Cabozantinib (XL-184, BMS-907351; Cabometyx®, Exelixis, Alameda, CA, USA) is a multi-kinase inhibitor targeting multiple TKs including MET. Patients with NSCLC with METex14 skipping alteration have shown the partial response to therapy after treatment with cabozantib.107,117 In a phase II study of solid tumors (ClinicalTrials.gov Identifier: NCT00940225) ORR was 10% and mPFS was 4 months.118 Importantly, some case studies of patients with NSCLC with METex14 alteration treated with cabozantinib showed intracranial response.117,119 Phase II studies in patients with NSCLC with MET deregulation are ongoing (ClinicalTrials.gov Identifier: NCT03911193 [CABinMET study], and ClinicalTrials.gov Identifier: NCT01639508).120
Merestinib
Merestinib (LY2801653) is another multi-kinase ATP-competitive inhibitor of MET.121 After the demonstration of an acceptable safety profile and potential antitumor activity in a phase I trial,122 a phase II clinical study (ClinicalTrials.gov Identifier: NCT02920996) is ongoing for the treatment of advanced NSCLC harboring METex14 alterations. A preclinical study demonstrated the antitumor response of merestinib in combination with emibetuzumab in a mouse model with METex14 skipping alteration.121 Recently, merestinib demonstrated antitumor activity in a patient with lung cancer harboring METex14 skipping and acquired resistance against capmatinib and crizotinib.123
Glesatinib
Glesatinib (MGCD265) is also a multi-kinase inhibitor of MET, AXL, VEGFR1/2/3, RON, and TIE2, which demonstrated antitumor activity in preclinical and clinical studies with METex14 alteration.109,124 A phase II study (ClinicalTrials.gov Identifier: NCT02544633) showed the antitumor activity of glesatinib in patients who had METex14 skipping alteration and acquired resistance against crizotinib.109,123,125
TPX-0022
TPX-0022 is a novel multi-kinase inhibitor of MET, CSF1R, and SRC, which demonstrated antitumor activity in preclinical xenograft models.126 A recent phase I clinical study reported that TPX-0022 was well tolerated, and responses were observed in patients with advanced solid tumors harboring genetic MET alterations (ClinicalTrials.gov Identifier: NCT03993873).127
Selective kinase MET inhibitor on METex14 skipping NSCLC
This group of inhibitors specifically target the MET receptor by binding to the ATP binding pocket of the MET kinase. Application of such selective MET inhibitors has produced a promising response in patients with METex14 skipping alteration. Capmatinib, tepotinib, savolitinib, and APL-101 are among the promising agents in this group.
Capmatinib
Capmatinib (INC280; TrabectaTM, Novartis, Basel, Switzerland) is a highly-selective, ATP-competitive MET inhibitor and the first and only MET inhibitor approved by the FDA to target metastatic NSCLC with METex14 skipping alteration as determined by an FDA-approved test.128–130 The approval is based on the results from the pivotal GEOMETRY phase II study (ClinicalTrials.gov Identifier: NCT02414139). The primary efficacy outcome based on ORR was 68% and 41% among 28 treatment-naive and 69 previously treated patients, respectively, based on the blinded independent review committee assessment. The median DOR was 12.6 months (n=19) for treatment-naive and 9.7 months (n=28) for pre-treated patients.131,132 Importantly, capmatinib also exhibited antitumor activity in patients with brain metastases in previously treated NSCLC harboring METex14 alterations.108,133
Tepotinib
Tepotinib (END 1214063; Tepmetko®, Merck KGaA, Darmstadt, Germany) is an ATP-competitive and highly-selective oral MET inhibitor, which showed MET inhibitory activity in in vitro and in vivo models. It supressed MET activation by both ligand-dependent and independent mechanisms.134 In March 2020, regulatory authority in Japan approved tepotinib for the treatment of NSCLC with METex14 skipping alteration.135 The approval was based on data from 99 patients with NSCLC with METex14 skipping alteration who had been followed up for 9 months in the ongoing single-arm phase II VISION study (ClinicalTrials.gov Identifier: NCT02864992). The primary endpoint ORR of the study, as assessed by an independent review committee, was 46% with median DOR of 11.1 months for patients identified by combined biopsy (liquid/tissue biopsy). The response rate was 48% for patients in the liquid biopsy group (n=66), and was 50% for those in the tissue biopsy group (n=60).89 The FDA granted tepotinib a breakthrough therapy designation for the treatment of NSCLC harboring METex14 skipping alterations in September 2019. Recently, a case of antitumor activity of tepotinib in a patient with NSCLC with brain metastasis harboring a MET gene rearrangement was reported.136
Savolitinib
Savolitinib (volitinib, AZD6094, AstraZeneca, Cambridge, UK) is also a highly selective MET inhibitor.137,138 Interim data from a phase II study (ClinicalTrials.gov Identifier: NCT02897479) reported encouraging antitumor activity and an acceptable safety profile of savolitinib in patients with METex14 skipping NSCLC, including pulmonary sarcomatoid and other histologies. The ORR from preliminary data (n=61) was 47.5% and mPFS was 6.8 months.139
APL-101
APL-101 (bozitinib, CBT-101, PLB-1001), another highly selective MET TKI, has demonstrated robust anticancer activity in various human xenograft tumor models with MET dysregulation and bears the potential to cross the blood–brain barrier in glioblastoma.140,141 Currently, SPARTA, a phase I/II study (ClinicalTrials.gov Identifier: NCT03175224) is evaluating antitumor activity of APL-101 in patients with NSCLC with METex14 skipping and solid tumors with MET aberrations.
Glumetinib
Glumetinib (SCC244) is a highly-selective, ATP-competitive MET inhibitor. The antitumor activity of this agent was demonstrated as equivalent to capmatinib in a preclinical study.142 Currently, phase I studies (ClinicalTrials.gov Identifier: NCT03466268) in patients with NSCLC with MET alterations, and another study (ClinicalTrials.gov Identifier: NCT03457532) in patients with solid tumors harboring MET alterations are ongoing for the evaluation of the safety and antitumor activity of glumetinib. A global phase I/II study (ClinicalTrials.gov Identifier: NCT04270591) for patients with NSCLC with MET alterations is also ongoing.
The effect of MET antibodies on METex14 skipping NSCLC
Emibetuzumab
Emibetuzumab (LY2875358) is a humanized bivalent anti-MET antibody that has high neutralization and internalization activities. It showed potent antitumor activity to inhibit HGF-dependent and HGF-independent tumor growth in mouse xenograft models and in MET–positive (including NSCLC) patients.143,144 A preclinical study revealed more antitumor activity of emibetuzumab combined with merestinib on gastric cancer with METex14 muation.121
Onartuzumab
Onartuzumab is another antibody drug that has shown antitumor activity in preliminary studies. However, it failed to improve the clinical outcomes of MET-positive patients compared with placebo in phase III studies.145
SYM015
SYM015 is a combination of two humanized antibodies directed at the elimination of the MET receptors.146 In a phase I/II study (ClinicalTrials.gov Identifier: NCT02648724), the safety and efficacy of Sym015 in patients with advanced NSCLC with MET amplification and exon 14 deletion were observed. Of 20 patients with NSCLC, the ORR was 25% and the disease control rate was 80%, with median PFS of 5.5 months.147
REGN5093
REGN5093 is a MET biparatopic antibody that blocks HGF binding and causes rapid internalization and degradation of MET.148 A phase I/II study (ClinicalTrials.gov Identifier: NCT04077099) demonstrated the safety and tolerability of REGN5093 in patients with NSCLC with MET alterations.149 This study is ongoing and is open for enrollment of patients.
The impact of antibody–drug conjugates on METex14 alteration
Telisotuzumab vedotin
Telisotuzumab vedotin (ABBV-399) is a conjugate of a MET-targeted antibody and monomethyl auristatin E. This antibody–drug conjugate has demonstrated antitumor activity in patients NSCLC with MET dysregulation in a preliminary analysis from a phase I study.150,151
Resistance of METex14 skipping alterations to MET TKIs
Reports suggest that patients with NSCLC with METex14 skipping alterations are sensitive to MET TKI treatment.87,107,111 However, emergence of primary or acquired resistance may challenge the efficacy of MET TKI-based monotherapy.11,123,157 Clinically, the analysis of pre- and post-MET TKI treatment data from 20 patients showed 35% on-target and 45% off-target resistance acquired after the treatment.123 Even though MET is exclusively a driver gene in many cancers, in some cases, METex14 skipping alterations may exist with alterations in other driver genes, such as amplifications of MDM2 (25–35%), CDK4 (3–21%), and EGFR (6–29%), leading to MET TKI resistance. Furthermore, mutations or amplification of KRAS (3–7%) and PIK3CA (3–10%), and loss of PTEN expression (23%) may exist with METex14 skipping alterations contributing to resistance.
The pre-existence of MET Y1230C on-target mutation in addition to METex14 skipping alteration has accounted for the primary resistance to crizotinib.158,159 MET-D1228N-acquired mutation was found to be responsible for the resistance to crizotinib in a patient with METex14 skipping alteration, who did not have any additional mutation in MET or other driver genes before the treatment started.11 A comprehensive analysis of secondary mutations using a Ba/F3 model resistance to eight TKIs reported that D1228 and Y1230 are common sites for resistance mutations for type I TKIs, whereas L1195 and F1200 are the mutations leading to resistance to type II TKIs. D1228A/Y accounts for resistance to both type I and II MET TKIs.160
In addition, tumor cells may activate other signaling pathways to counterbalance the MET TKI suppressed signaling. In such cases, alterations leading to overexpression of key proteins drive the activation of alternative receptors, which leads to the sustained activation of major signaling pathways (bypass signaling) and contributes to the therapeutic resistance, regardless of effective MET inhibition by MET TKI drugs. On the other hand, MET dysregulation, mostly due to METex14 skipping alteration, has also been observed in NSCLC tumors because of off-target acquired resistance to EGFR TKIs.161
Identification of the resistance mechanism to MET TKIs is crucial for the effective treatment of NSCLC. For example, tumors developing acquired resistance against glesatinib (type II) through the amplification of the mutated METex14 allele showed partial response after switching to crizotinib (type Ia).160 Merestinib (type II) was reported to function better against D1228N–mediated acquired resistance, which developed after the application of capmatinib (type Ib).109 Similarly, glesatinib (type II) revealed better antitumor activity in Y1230H/S mutation, which developed after crizotinib (type Ia) treatment.123 Preclinical and clinical data have demonstrated that tumors harboring METex14 skipping alterations along with other mutations, RAS MAPK or PI3K/AKT pathway mutations, show reduced response to MET TKIs.162–164 Overall, the resistance mechanisms against different types of MET inhibitor can be different.109 Therapy combining relevant inhibitors can be helpful for the treatment of NSCLC with METex14 skipping alteration along with other driver mutations.164,165 Extensive studies are needed to unravel the full spectrum of resistance mechanisms against the inhibitor drugs for optimising therapeutic intervention.
Conclusion and outlook
MET abnormalities due to METex14 skipping alteration can drive cancer by upregulating receptor activity. It has become a promising target for kinase inhibitor-based targeted therapy in NSCLC, and several recent clinical trials have demonstrated the strong therapeutic effect of such inhibitors. However, it is not yet available to many potential patients who could benefit from such therapy. Firstly, current guidelines do not necessitate the analysis of METex14 skipping alterations for a standard treatment plan, missing the identification of such mutations in many patients and consequently precluding a possible target group from receiving MET inhibitors. Secondly, many early-stage patients may not be subjected to molecular profiling due to the complexity in accessing the tissue biopsy sample. In such cases, application of liquid biopsy and ctDNA genotyping can ease sample availability, which can widen the scope of diagnosis, thereby enabling more patients to get a proper diagnosis and therapy. In addition, a detailed understanding of primary and acquired resistance mechanisms can aid in decision making for the appropriate therapeutic intervention. More clinical trials focusing on the combination of MET inhibitors with inhibitors of other signaling pathways can help to identify an appropriate drug combination in MET inhibitor-resistant cancers.