Over the course of the last three decades there has been significant improvement in the outcomes of children with non-Hodgkin lymphoma (NHL).1 However, despite varied treatment strategies and sequential clinical trials, the prognosis for one NHL subtype, anaplastic large cell lymphoma (ALCL), has remained largely unchanged.2 ALCL, a peripheral T-cell lymphoma, was first described in 1985 by Stein et al., and accounts for 10–15% of all pediatric NHL.2,3 Approximately one-third of children treated with modern multi-agent chemotherapy regimens will have disease recurrence and one-half of those will ultimately die of their disease. The incorporation of novel targeted therapies such as anti-CD30 antibody drug conjugates and anaplastic lymphoma kinase (ALK) inhibitors into therapy for children with ALCL provide hope for improvement in these outcomes.2,4–8
This review article will focus on the pathology, clinical presentation, prognostic factors, and therapy, including new agents, for ALCL in children.
Pathology
Anaplastic large cell lymphoma is a peripheral T-cell lymphoma, characterized by strong expression of CD30. There can be variable expression of T and natural killer (NK) cell markers, with less than 5% of childhood cases being assigned a null-cell phenotype.9 Most cases express epithelial membrane antigen and, unlike adult ALCL, the vast majority of pediatric cases (>95%) demonstrate overexpression of ALK.10 The classic (2;5) translocation of ALK with the nucleophosmin gene (NPM) is seen in approximately 85% of cases.11 As NPM encodes a nucleolar domain, ALK staining occurs in both the cytoplasm and nucleus. The presence of only cytoplasmic ALK staining is suggestive of a variant ALK translocation.11
The histology of ALCL was originally described as exhibiting sheets of large pleomorphic blasts with horseshoe-shaped nuclei and multiple nucleoli (hallmark cells). ALCL is classified into five histologic subtypes: common/classic, small cell, lymphohistiocytic, Hodgkin-like, and composite.12 In the largest pediatric study to date, ALCL 99, the common/classic subtype accounted for 65% of cases, and the composite subtype accounted for 25% of cases. 9 The small cell variant contains predominantly small cells with irregular nuclei and CD30 staining that is perivascular.13–15 The lymphohistiocytic subtype contains a dominant population of reactive histiocytes that, unlike the tumor cells, are CD30 negative (-) and Ki-67-. The Hodgkin-like variant contains multi-nucleated cells with a Reed-Sternberg appearance, while the composite variant contains more than one pattern within the same specimen.13–15 Despite the increasing use of molecular testing, the histologic characterization of ALCL remains important as the small cell and lymphohistiocytic subtypes have been associated with an increased risk of relapse.9
Clinical presentation and staging
The mean age of presentation for ALCL is 12 years, and is rare in children of less than one year of age.2,16 ALCL affects boys more than girls at a ratio of 6.5:1.15,17 The most common presenting features include lymphadenopathy and systemic symptoms, such as fever and weight loss; however, ALCL can also present with an unusual course of waxing and waning lymphadenopathy or other symptom constellations, which may confuse the initial diagnosis.2,18 ALCL often presents with advanced stage disease and in extra-nodal locations, such as skin and bone.2,12,15 In 12% of patients, the presentation of ALCL will be complicated by features of concomitant hemophagocytic lymphohistiocytosis (HLH). Lung, bone marrow, and central nervous system (CNS) involvement has been found to be more common in patients presenting with HLH-associated ALCL, although the outcomes are similar to those without HLH.19
The diagnosis of ALCL requires a biopsy of affected tissue, nodal or extra-nodal. Samples are required to be adequate for morphologic and molecular analysis, and therefore fine needle biopsies are discouraged as they often do not provide adequate tissue for evaluation. The staging evaluation includes physical examination, computed tomography (CT) scans or magnetic resonance imaging (MRI), and assessment of bone marrow (BM) and cerebrospinal fluid (CSF). In addition, [18F]fluorodeoxyglucose (FDG)-positron emission tomography (PET) scanning may be helpful if available. While FDG-PET modalities have been incorporated into the Lugano classification for NHL in adults, there are minimal data on its use in children with NHL.20,21 Therefore it has not been incorporated into the staging classification for pediatric NHL.
The St Jude Childhood and Adolescent NHL staging system, first developed in 1980, forms the basis for the Revised International Pediatric NHL Scoring System (IPNHLSS), which incorporates information regarding nodal and extra-nodal sites of disease and molecular testing of bone marrow and CNS sites.20 CNS involvement is defined as any CNS tumor mass, cranial nerve palsy that cannot be explained by extradural lesions, or blasts in the CSF. Cerebrospinal fluid positivity is based on morphologic evidence of lymphoma cells and should be considered positive if any number of blasts are detected.20 Bone marrow involvement is defined as evidence of ≥5% blasts or lymphoma cells on bone marrow aspiration. Bone marrow and CNS classifications are evolving rapidly; however, as the use of minimal disseminated disease (MDD) measurements are incorporated into clinical practice, they are not yet used within the official staging classification.20
Prognostic factors
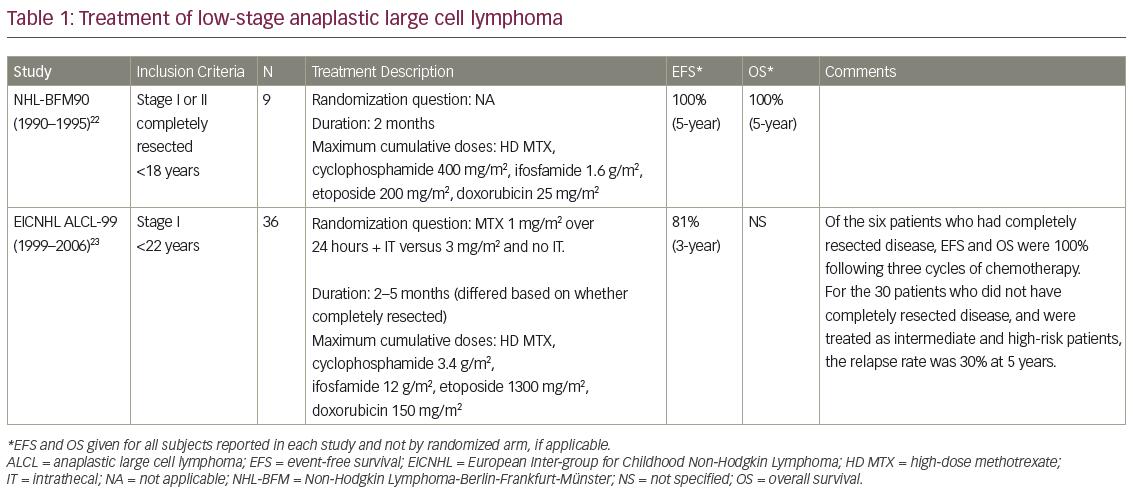
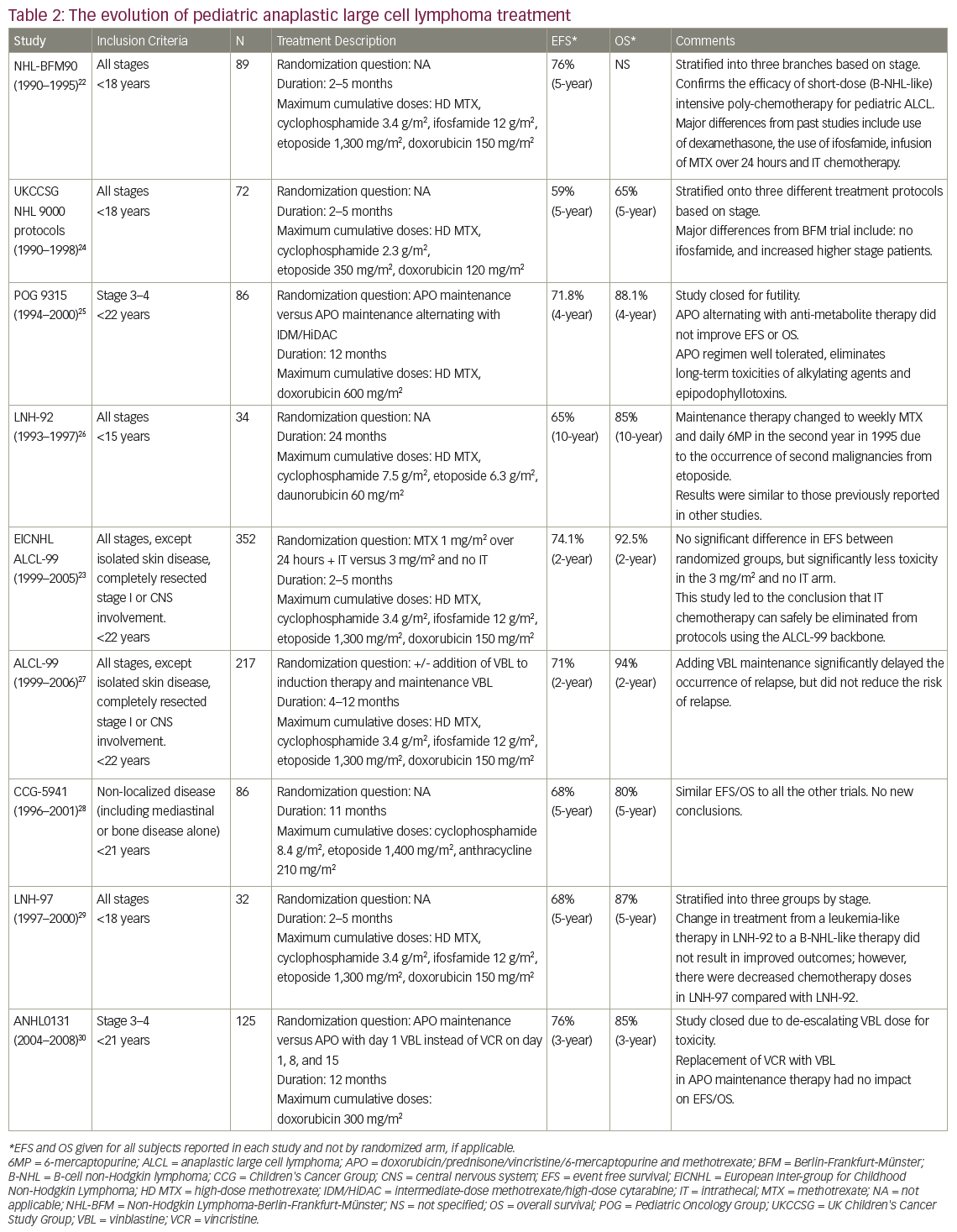
The utility of clinical features to predict risk of relapse in ALCL remains questionable. The major co-operative groups treating pediatric patients with ALCL have identified a number of risk factors within individual trials, although none have consistently been shown to be prognostic across trials. Major pediatric trials in ALCL are further described in Tables 122–3and 222–30. The Berlin-Frankfurt-Munster (BFM) 90 study found the presence of B symptoms to be a risk factor for disease relapse whereas the Children’s Cancer Group (CCG) 5941 study described bone marrow involvement as prognostic.22,28 The European Intergroup Study for ALCL attempted to overcome the challenge of individual studies each with small patient numbers by combining data from the BFM, the French Society of Pediatric Oncology (SFOP) and the United Kingdom Children’s Cancer Study Group (UKCCSG) studies between 1987 and 1997.31 This analysis suggested mediastinal, visceral, and skin-involvement as high-risk features. However, results from ALCL 99, the largest prospective study containing 352 patients, did not support these findings.9
ALK negative (-) ALCL has been shown to be a negative prognostic factor in adult ALCL.17 However, as the overwhelming majority of pediatric patients with ALCL are ALK positive (+), the prognostic significance of ALK-disease in childhood remains unclear. Histologic subtype has been shown to be associated with increased relapse risk in several studies. For example, the ANHL 0131 study found small cell variant histology to be associated with a significantly worse 3-year event-free survival (EFS) and overall survival (OS) compared with the common histology (hazard ratio [HR]: 14.3, p<0.0001).30 Similarly, despite an entirely different chemotherapy backbone, ALCL 99 found both small cell and lymphohistiocytic histologies to be associated with an increased risk of relapse (HR: 2.0, p=0.002).9 In both studies, the small cell variant histology accounted for approximately 30% of patients.
In addition, the use of molecular techniques to detect MDD and minimal residual disease (MRD) has been shown to be of prognostic value.32–3 Damm-Welk et al. identified the presence of the NPM-ALK fusion transcript in 42% of patients who had morphologically negative bone marrow at diagnosis (MDD).32 These patients had a cumulative incidence of relapse (CIR) of 50%, compared to 15% of those who were MDD-. Similarly, the persistence of the NPM-ALK fusion transcript after the first cycle of therapy (MRD) also portends a high risk of relapse. Damm-Welk et al. demonstrated that patients who were MDD+/MRD+ had a CIR of 81% (n=26), compared to 31% for those who were MDD+/MRD-, and 15% for the group that was MDD-.34 Together, these studies suggest that the use of MDD and MRD can identify the patients at highest risk, and may allow for response-based therapy in future studies.
Immune response to ALCL, measured in anti-ALK antibodies, has been shown to correlate with prognosis. Ait-Tahar et al. identified ALK antibodies in 87 of 95 patients tested prior to the start of therapy. There was a significant inverse relationship between antibody titre and risk of relapse.35 Subsequently Mussolin et al. described risk stratification of patients by combining antibody response and MDD.36 A total of 128 patients treated on the ALCL 99 protocol were divided into high risk (MDD+ and low antibody titre, <1/750), low risk (MDD- and antibody titre >1/750), and intermediate risk (all other patients) groups. Progression-free survival was 93%, 68%, and 28% for low, medium, and high risk groups, respectively.36 Further prospective studies are needed to verify such risk stratification.
Treatment of anaplastic large cell lymphoma
A variety of treatment strategies have been used in the treatment of ALCL over the last three decades, with different protocols utilised by European and North American groups. The BFM and Italian groups have developed shorter but more intensive regimens, analogous to B-NHL therapies. These regimens are generally administered on an inpatient basis and involve higher doses of alkylating agents while avoiding anthracyclines.22,27,37,38 By contrast, the CCG, Pediatric Oncology Group (POG) and now the Children’s Oncology Group (COG) in North America have developed protocols similar to those typically used in the treatment of leukemia.25,28,30 The leukemia-like protocols have a longer duration and are administered largely on an outpatient basis. The protocols include higher cumulative anthracycline exposure but with less or no alkylating agents.4,25,27–8,30,37–8 Despite the differences in agents, therapy duration and dose intensity, the outcomes for patients have largely remained unchanged, both between regimens and across time (see Tables 1 and 2).
Patients with low stage ALCL are relatively rare. Children with low stage disease, especially those with completely resected disease, have been shown to have excellent outcomes with short courses of combination therapy (see Table 1). The BFM group reported a 5-year EFS of 100% for nine patients with completely resected stage I and II disease with three cycles of chemotherapy. By contrast, the 5-year EFS was 73% for non-resected stage II and stage III disease with six courses of inpatient chemotherapy.22 In a report on 36 patients with stage I disease in the ALCL 99 study, those who had completely resected disease and received short-course chemotherapy had a 2-year EFS of 100% whereas those with incompletely resected disease had a 2-year disease-free survival (DFS) rate of 30%, which was similar to patients with higher stage disease.23
Table 2 describes nine co-operative group trials completed since 1990 for children with all stages or advanced stages of ALCL. The most recent European trial, at the time of writing, was the aforementioned ALCL 99 study. The chemotherapy backbone used for this trial was similar to that of the BFM 90 protocol, and included a 5-day prophase followed by six alternating courses of intensive chemotherapy.27,37 The study attempted to answer two experimental questions through two randomizations. The first question was whether the addition of vinblastine to the regimen, and as a year of weekly maintenance therapy, affected EFS. The addition of vinblastine was found to delay disease relapse, but after 2 years of follow up, no significant difference was identified between the two arms (EFS of 72% versus 70%, p=0.71).27 The second randomization involved treatment with methotrexate (1 mg/m2 over 24 hours) with additional intrathecal methotrexate on day 1 of each cycle compared with methotrexate (3 mg/m2 over 3 hours) with no additional intrathecal methotrexate. The 2-year EFS did not differ between arms (73.6% and 74.5%, respectively; HR: 0.98; confidence interval [CI]: 91.76%, 0.69–1.38).37 Of note, the risks of grade 4 hematologic toxicity (79% and 64%, p<0.0001), infection (50% and 32%, p<0.0001) and grade 3–4 stomatitis (21% and 6%, p<0.0001) were all significantly increased in the 1 mg/m2 methotrexate group.37 In summary, the ALCL 99 trial demonstrated that the addition of vinblastine did not improve EFS, and that higher dose but shorter infusion duration of methotrexate with no intrathecal therapy provided the same 2-year EFS with less toxicity.27,37
The most recently completed COG trial, titled ANHL0131, used the APO chemotherapy backbone (vincristine, adriamycin, and prednisone), which includes induction therapy followed by 15 cycles of maintenance therapy. In contrast to the ALCL 99 protocol, this regimen includes intrathecal chemotherapy and a cumulative anthracycline dose of 300 mg/m2, but no alkylating agents. Patients in this study were randomized to receive either vincristine, which was part of the standard APO regimen, or vinblastine with each cycle.30 The 3-year EFS and OS between study arms did not exhibit statistically significant differences (79% versus 74%, p=0.68, and 86% versus 84%, p=0.87, respectively).30 There was a significantly higher incidence of neutropenia and infection in the group treated with vinblastine (84% versus 39%, p<0.0001, and 43% versus 22%, p=0.019, respectively).
The currently enrolling COG trial, ANHL12P1, is a safety and feasibility trial incorporating two novel agents, brentuximab vedotin and crizotinib, into the ALCL 99 backbone. The decision to adopt the ALCL backbone for this study was made with consideration of lower cumulative anthracycline dosage and shorter duration of therapy, as well as the opportunity to attempt to replicate data for the prognostic value of MDD and MRD in the context of this therapy.
Relapsed or recurrent disease
There is no consensus on the best treatment for relapsed or refractory ALCL. Protocols have varied from single-agent chemotherapy to multi-agent reinduction chemotherapy consolidated by autologous stem cell transplant (SCT) or allogeneic SCT.
Prognostic factors in relapse
Several risk factors have been associated with unfavourable outcomes in retrospective case series. Early relapse, occurring within 12 months from diagnosis, has been the most consistently identified poor prognostic factor.39,40 In the study by Brugières et al., patients with early relapse had a 3-year EFS of 28% (n=24), as compared with 68% (n=17) for those who relapsed more than 12 months after diagnosis (p=0.01).39 In the report by the BFM group, patients with bone marrow or CNS involvement had a 5-year OS of 27% (n=11) compared with 62% for the patients who did not (n=63, p=0.001).40 This study also implicated CD3 positivity as a high-risk factor, particularly for patients treated with autologous SCT (5-year EFS 18% compared with 72% for CD3- patients, p<0.001).
Therapy for relapsed and recurrent disease
Brugières et al. reported the activity of single agent vinblastine in a series of 36 patients with relapsed refractory ALCL.41 Of the 30 patients they were able to evaluate, in 83% (25/30) vinblastine induced a complete remission (six patients could not be evaluated as the entire tumor was excised). While this was a retrospective series with a heterogeneous group of patients, there were two additional observations. Firstly, nine of the 25 patients who achieved complete remission (CR) remained in long-term remission with a median follow up of 7 years. Secondly, of the 19 patients who developed a subsequent relapse, 12 were again treated with vinblastine, and 11 of these patients achieved a repeat CR. The OS for this multiple-relapsed group was 65%, suggesting that recovery was possible.
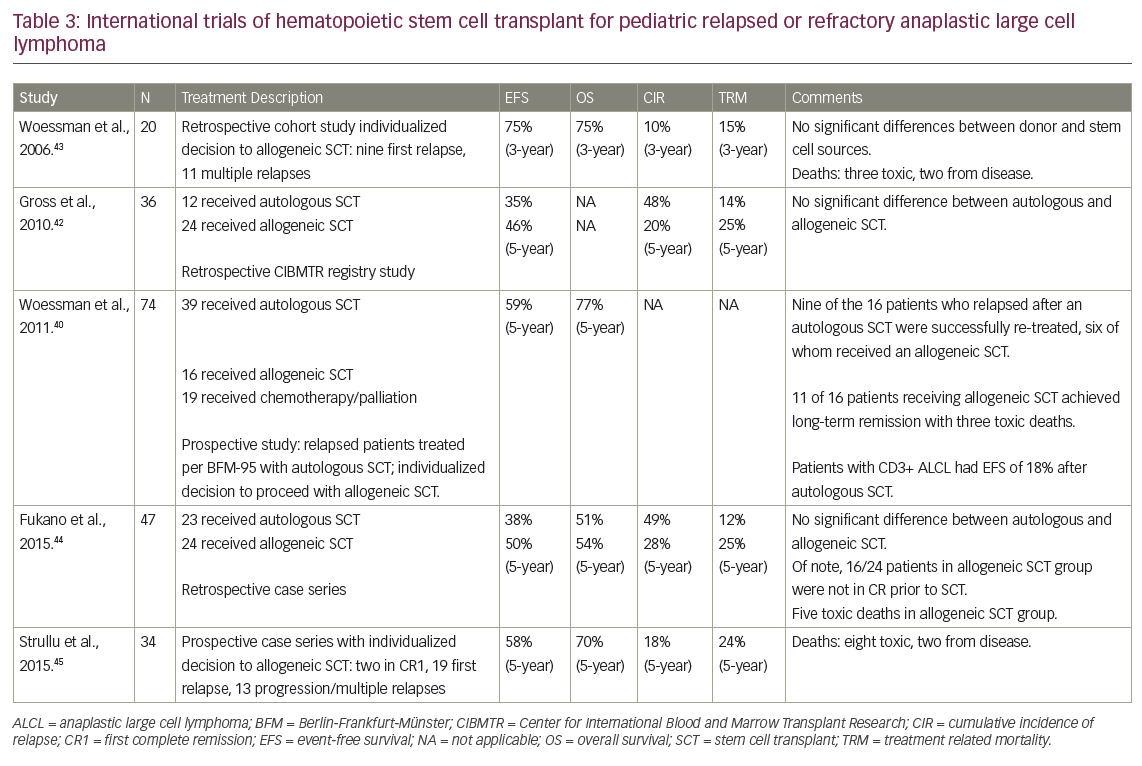
To the best of our knowledge, as yet there are no published prospective studies comparing consolidation with an allogeneic SCT or autologous SCT following high-dose chemotherapy. In addition to heterogeneity in the high-dose chemotherapy used pre transplant, patients with perceived higher-risk relapse are more likely to be treated with allogeneic SCT, making comparisons with autologous SCT inherently biased. Nevertheless, a number of retrospective series have suggested that allogeneic SCT may be superior.40,42–3 However, allogeneic SCT has been consistently associated with high rates of toxicity, with rates of transplant-related mortality measured at around 25%.40,42–3 These studies are summarized in Table 3.40,42–45
The European Inter-Group for Childhood Non-Hodgkin Lymphoma (EICNHL) has implemented a prospective ALCL relapse trial, the results of which have been published only in abstract form.46 They used a risk-adapted approach with patients divided into four arms. Patients with progression during first-line therapy (arm one) and those with CD3+ relapse after initial therapy (arm two) were eligible for allogeneic SCT. Patients with CD3- relapse with 12 months of diagnosis (arm three) were consolidated with autologous SCT after BEAM chemotherapy (carmustine, etoposide, cytarabine, and melphalan), and those with CD3- relapse more than 12 months from diagnosis were treated with vinblastine weekly.46 The 3-year EFS for patients who received allogeneic SCT (arms one and two) was 64%, with patients in arm one having an EFS of 41% (n=19) and those in arm two having an E FS of 81% (n=26). While the full study is yet to be published, the data suggest that allogeneic SCT is an effective therapy for patients with high-risk relapsed ALCL.46 Conversely, patients with early CD3- relapse had a 3-year EFS of only 35% (n=31), suggesting that autologous SCT is not as effective. Encouragingly, the reported 3-year EFS in low-risk relapsed ALCL treated with single agent vinblastine (arm four) was 85%.46
Together, these studies demonstrate that relapsed ALCL is a chemosensitive disease of which some patients with low-risk relapses can be treated with vinblastine monotherapy as a curative agent, whereas those high-risk patients should be considered for allogeneic SCT. The role of new agents in the relapse setting is an area of active investigation.
Novel agents
Brentuximab
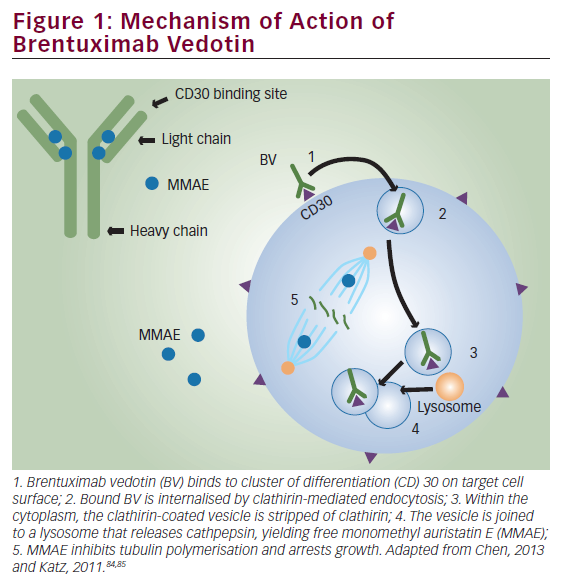
Cell surface expression of CD30 is a hallmark of ALCL. Brentuximab vedotin is an anti-CD30 antibody conjugated to monomethylauristatin E (MMAE), an anti-microtubule agent. When brentuximab binds to CD30, the complex is internalized, and the released MMAE disrupts microtubules and promotes cell death by apoptosis.15 Pro et al. reported a phase II study in adults with relapsed or refractory ALCL in which brentuximab was administered at a dose of 1.8 mg/kg every 3 weeks.7 A total of 50 out of 58 patients (86%) achieved an objective response, with 57% achieving CR and 97% exhibiting tumor reduction. This cohort of patients had particularly high-risk disease, with 62% having primary refractory disease. These data led to approval of brentuximab in adult patients with relapsed ALCL by the US Food and Drug Administration (FDA).7 A follow-up study reported 5-year PFS of 39% and 5-year OS of 60% in the same cohort with the majority of patients receiving an allogeneic SCT after brentuximab.47 Notably, eight of the 38 patients who achieved CR with brentuximab remained in sustained long-term remission without any additional therapy (median observation time: 75 months). The most commonly seen toxicity was peripheral neuropathy (57%), of which two-thirds resolved completely over the follow-up period.47 There are case reports of patients with CD30+ ALCL treated with brentuximab who have subsequently relapsed with CD30- disease, suggesting a potential mechanism of failure.48–50
Safety of single agent brentuximab has been studied in children with relapsed and refractory Hodgkin lymphoma and ALCL.51 The safety and feasibility of the addition of brentuximab to the ALCL 99 regimen is currently being assessed in a phase II study of children with newly diagnosed ALCL through the Children’s Oncology Group (ANHL 12P1).
ALK inhibitors
Crizotinib is a small molecule, competitive ALK and hepatocyte growth factor receptor (MET) kinase inhibitor that blocks the activity of the NPM-ALK fusion protein.52 ALK inhibitors, including crizotinib, have been studied in a number of ALK+ adult malignancies. For example, in ALK+ non-small cell lung cancer (NSCLC) response rates in previously chemo-refractory patients have been shown to be higher with the use of ALK inhibitors. This has led to the adoption of ALK inhibitors as part of the adult standard of care when ALK mutations are identified.53–4 The adult recommended dose is 250 mg orally twice daily (or approximately 140 mg/m2).55 Crizotinib is well tolerated with minimal side effects. Reported side effects include nausea and vomiting, and visual disturbances during the first cycle, as well as ongoing bone marrow suppression (rarely grade 4 neutropenia).52,55
The safety and efficacy of crizotinib has been studied in children with ALK+ neoplasms including neuroblastoma, inflammatory myofibroblastic tumors and ALCL. ADVL 0912, a phase I study investigating the pharmacokinetics of crizotinib in children with solid tumors and ALCL, found that the pharmacokinetics in children are similar to those in adults.55 The maximum tolerated dose in children was found to be 280 mg/m2; however, a dose of 165 mg/m2 was also found to be effective, with a similar side effect profile to that observed in adults.52,55 Of the eight patients with ALCL who had radiographically measurable disease, six had a complete response, one had a partial response and one had stable disease.52 In the extension phase II component of the crizotinib study, 22 of a total of 23 patients were found to have a reduction in transcript levels of ALK using polymerase chain reaction within one month of the start of treatment, and 13 out of those 22 had a 75% reduction or more in the transcript levels.6 Grade 3 and 4 toxicities were noted in 33% of the 165 mg/m2 group and 85% in the280 mg/m2 group, with 70% of the toxicities attributable to neutropenia.6 A total of 12 of the 16 patients who came off therapy with crizotinib proceeded to transplant, two patients continued crizotinib off study and two refused further treatment.6
Many questions regarding the use of crizotinib in pediatric patients with ALCL still exist, including the appropriate duration of therapy and potential for discontinuous use. Early relapse following the discontinuation of crizotinib has been observed in both children and adults, and the average length of treatment in the studies so far is variable.56 It is possible that the treatment may need to be used on an ongoing maintenance-like basis. There is also preliminary evidence that discontinuation of crizotinib may be effective. Using in vivo mouse models, Amin et al. found that drug-resistant ALCL cells upregulated the production of ALK in response to the use of ALK inhibitors.57 When the ALK inhibitor was discontinued in these cases, the excess ALK signaling rapidly arrested or killed the cells, causing ongoing tumor control.57 Further study is needed to determine the optimal use of ALK inhibitors.
The safety and feasibility of adding crizotinib to intensive multi-agent chemotherapy is being studied in patients with newly-diagnosed disease by the COG. In addition, the Pediatric MATCH trial is investigating targeted therapies directed by genetic testing in pediatric patients with relapsed or refractory solid tumors, NHLs or histiocytic disorders. For those patients with ALK targetable lesions enrolled in this trial, treatment with crizotinib will be investigated. ALK inhibitors are yet to be tested in combination with anti-CD30 therapy.
There are additional ALK inhibitors; however, for the majority of them there is very limited evidence for their use in pediatric cases. Ceritinib and alectinib, like crizotinib, are currently approved for use in adult patients with ALK+ NSCLC.56,58–9 They have been shown to be clinically active in crizotinib-resistant ALK+ NSCLC, as they are not affected by the same bypass routes used by the resistant tumor cells to avoid the effects of crizotinib.58,60 Other next generation ALK inhibitors include brigatinib, and PF-06463922.61–2 This list will continue to increase as more drugs are developed.63–4
Given the relative rarity of pediatric ALCL, the investigation of these new agents in children will require ongoing co-operative group protocols and novel study designs.
Conclusion
With conventional combination chemotherapy, the majority of children with ALCL can be cured of their disease. Over the last several decades, improvement in outcomes has been frustratingly slow despite sequential co-operative group clinical trials. It is hoped that the incorporation of targeted therapies such as brentuximab and ALK inhibitors will be key components in substantially improving outcomes for those children with currently difficult-to-cure disease.