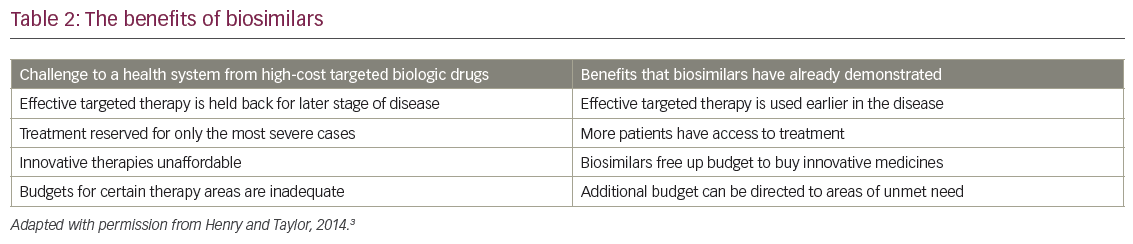
Biosimilars, which are copies of patent-expired large-molecule biologic drugs (Table 1),1–8 offer the same rationale as generic drugs. They can give patients, hospitals and healthcare systems the opportunity to expand patient access to care and offer potential budget savings to reinvest (Table 2).1,2,9 They are designed to be similar in effectiveness, safety and quality to an original ‘reference’ drug. As a result, their impact is not clinical, but primarily economic. By offering competing brands of the same drug they can create price reductions for both biosimilar and original reference drugs; savings in price compared to the reference drug have been as great as 72% when transparent tenders for drugs have been reported.10
Biosimilars have been used to treat patients in Europe for a decade and have recently been introduced in the US. Patents are due to expire for a large number of biological cancer drugs by 2020, which opens the market to more biosimilars.11 Harmonisation between the European and American clinical and regulatory requirements for biosimilars is increasing, which will aid the licensing of these drugs, but there are considerable differences in both pricing and uptake around the world. This is probably due to variations in post-market policies.
Economic rationale for biosimilars
Biologic medicines are transforming cancer by targeting the key pathways that underlie mechanisms of disease. In many instances, they offer treatment with higher activity and lower toxicity than the previous generations of cancer treatment. However, biologics are more expensive to manufacture than the previous generation of small-molecule drugs and current requirements for registration studies add to the cost. This partly explains why cancer drug prices are rising five times faster than other areas of medicine and why oncology is such a focus for cost control policies.12,13 Since biologics are complex molecules produced by living cells, it is impossible to develop an identical biosimilar, as is the case with small-molecule drugs (generics).14 In addition, compared with first-generation biosimilars (e.g. growth factors, hormones), the evaluation of monoclonal antibodies (MAbs) is more problematic as they have a significantly increased structural complexity and several potential modes of action. Furthermore, they are generally administered in combination with other drugs, thus comparative clinical trials are challenging.15
With rising costs of treatment, the ageing of the world’s population and increasing global cancer incidence, there is a worry that cancer care is becoming unaffordable even in the richest countries. This is especially true in Europe, where Article 168 of the Treaty on the Functioning of the EU and Article 35 of the Charter of Fundamental Rights of the EU includes ‘the right to benefit from medical treatment…regardless of financial means, gender or nationality.’16 In the US too, there is concern as cancer drugs currently comprise eight out of the top ten most expensive drugs covered by Medicare.17
Cost can no longer be ignored by any country. The global spend on drugs for cancer was $79.2 billion in 2014; this is expected to grow by 11.6% per year with a predicted requirement for a budget of $153.1 billion by 2020.18 Drug costs now impact directly on the treatment choice.19–21 The choices advocated by oncologists (including, but not limited to, anticancer and supportive care drugs) regularly influence cancer care costs.22 Oncologists therefore need to offer leadership in cost control both for individual patients and for society.

There is a danger that, with rising costs for patients and payers, effective supportive care may be withheld, rationed or declined by patients in favour of spending on active cancer therapies. This would particularly affect expensive drugs such as haematological growth factors.23
Some costs of cancer care have been checked to date by the use of generic drugs.24,25 These are copies of small-molecule drugs that have lost patent protection. They reduce the cost of medicines and benefit healthcare systems by enabling the same budget to treat more patients.26 Generic drugs also deliver budget savings that can be reinvested in the next generation of innovative medicines. These savings from lower cost drugs that are introduced after loss of patent exclusivity by original reference drugs are significant. The National Health Service in England and Wales saved £12.3 billion (€19.9 billion/$15.1 billion) in 2013 through use of generics,27 and German insurance funds saved €12.9 billion in 2011 ($15.9 billion).28 Generic products saved the US health system nearly $1.5 trillion (€1.1 trillion) over the past 10 years (2004–2013) and $239 billion (€181.5 billion) in 2013 alone.29 For a hospital drug budget, generics will deliver the greatest value in two areas: oncology drugs and systemic anti-infective drugs.30 A clear example of the benefit of lower generic prices was the trends seen after the approval of generic carboplatin in the US. Total cost of carboplatin fell from $74 million to $36.5 million while at the same time monthly product sales in milligrams increased.31 However, even in rich European countries and the US, access to healthcare is limited and often access to new oncology medicines is low.32
Some countries have made great use of generics to increase access to care, but this has not been universal. Worldwide in 2011, generics accounted for about three-quarters of the volume of pharmaceuticals covered by basic health coverage in Germany, the UK, New Zealand and Denmark, while they represented less than one-quarter of the market in Luxembourg, Italy, Ireland, Switzerland, Japan and France.33
Since 2007, Europe has had access to two key supportive care cancer drugs in biosimilar versions: granulocyte colony-stimulating factors (G-CSF) filgrastim and epoetin-alfa. Examining the impact of biosimilar G-CSF filgrastim and epoetin-alfa allows us to assess the effectiveness of the biosimilar regulatory strategy that oversaw their development and their impact on patient access through cheaper drugs.
A biosimilar regulatory pathway can resemble a manufacturing change process for a reference biologic product.34–36 In one example of a manufacturing change to a European Medicines Agency (EMA)-approved biologic reference medical product, darbepoetin underwent re-establishment of master cell bank, modification of the vector used to produce the antigen/source material (including a new master cell bank), a change from roller bottle manufacturing process to a more scalable high-throughput (HT) process using cells in suspension, and a change of cell culture medium.37 To justify that the drug maintained its authorisation for primary and extrapolated indications, the manufacturer performed an animal pharmacokinetic (PK) study, an animal multiple-dose safety and PK/pharmacodynamic (PD) study, a phase I comparative randomised two-way study, an open-label crossover PK study with two single doses in 48 healthy volunteers, a phase III comparative efficacy study, a randomised controlled study of 446 patients in one indication and a single-arm open label safety study with an HT product in 1,172 patients in one indication.37 In the EMA assessment report, the change was rated as ‘replacement of a biological substance or product of biotechnology with one of a slightly different molecular structure’. Interestingly, the authors know of no concern raised over this degree of manufacturing change by medical societies in meeting presentations, editorial or comment papers, or representations to regulators. This stands in contrast to the case for biosimilar drugs; frequent public interventions and ‘position statements’ raise awareness and address concerns that slightly different molecular structures could represent an unacceptable risk.38–46 A ‘position statement’ is defined as describing only one side of an arguable viewpoint.47 Position statements may not, therefore, be the best way to review such an important topic that addresses both healthcare costs and access to innovative treatment. A structured search of the worldwide web using the exact terms ‘position statement biosimilar’, in November 2015, identified more than 3,000 unique associated web pages.48
One key reason to review biosimilar filgrastim and epoetin is to investigate whether the World Health Organisation (WHO) requirements of similar quality, safety and efficacy of biosimilar medicines has been achieved in practice. The safety of switching patients from one brand of drug to a biosimilar will be important in this as some commentators have suggested that anti-drug immune reactions could be created.49 Drug regulators intend that there are no clinically meaningful differences between biosimilars and the original refence drugs. If that is confirmed in practice, the success of biosimilars will need to be shown through primarily economic endpoints: impact on price, cost minimisation of the health budget and increased patient access to reimbursement for effective treatment.
Switching between reference and biosimilar products should in theory have no differences in risk than using different batches of the same reference drug over time in the same patient.50 This is because both manufacturing processes will never make ‘identical’ chemical structures over time and a biologic drug could never be a ‘generic’ copy of itself. This is firstly because many drugs are a mixture of different isoforms of the recombinant protein and show a natural variation in structure.51 Secondly, most reference drugs will undergo many changes to the modifications in manufacturing process, formulation or primary container over time. These changes in manufacturing can induce changes in the product attributes that can have an impact on clinical safety and efficacy.52
Changes to the formulation are done for very good reasons such as scaling-up production to meet higher sales, switching production to different factories and increasing the yield or purity of the drug. It is vital to minimise the variation in structure to ensure the final product has no clinically meaningful differences in quality, efficacy or safety; indeed, there is international harmonisation in regulatory analytic standards to control these processes through comparability ‘exercises’ of the drug before and after the formulation change.53 The comparability protocols for changes in reference drugs almost never involved clinical trials and they have never required switching safety studies between pre- and post-change product. If a product is judged to be comparable pre- and post-manufacturing change, then the drugs resulting from the process before and after the change are deemed interchangeable.52 Although there have been very rare incidences of complications that follow these steps, there are no suggestions that this is not a well-accepted process.54,55 Indeed, it was from this successful process for controlling manufacturing changes that the biosimilar regulatory pathway was developed.52
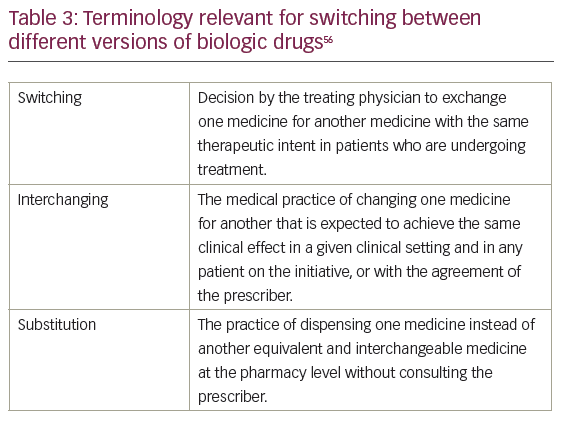
Crucial to the discussions on switching drugs is the terminology used (Table 3).56 This paper follows the convention described in the European Commission Consensus document ‘What you need to know about biosimilar medicinal products’.56
It is also an opportunity to test the capabilities of the ‘EudraVigilance’ European Drug Adverse event reporting system, to see if the lack of unique International Non-proprietary Names (INN) for EMA-approved biosimilars means that unexpected toxicities cannot be traced back to the specific version of a biologic drug.57 Within the EU there is mandatory prescribing of all biologic drugs by brand name, as well as recording of batch number of the product used.58
Post-approval studies can be crucial for payers in assessing the roles of biosimilars. As many regulatory biosimilar trials are based on surrogate endpoints, such as white cell count or haemoglobin (Hb) concentration, clinical health outcome data may be needed. Such data would show rates of clinically important outcomes such as neutropenic fever, hospitalisations and transfusions. Furthermore, effectiveness of a drug in routine use may be different in a real-world rather than in a clinical trial setting.59
The current impact of biosimilars in supportive cancer care
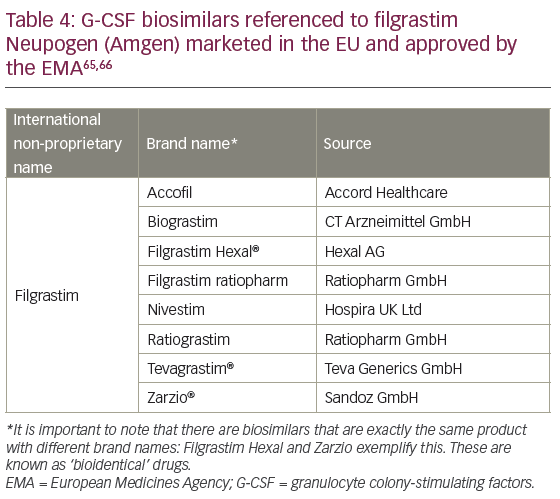
Supportive cancer therapy is the use of medicines to counteract unwanted effects of cancer treatment.60 Therapies for supportive care were the first approved biosimilars, represented by filgrastim and epoetin alfa, in the EU in 2007.61 Since then many development programmes for biosimilars have been initiated, such as for rituxumab, trastuzumab and bevacizumab. Filgrastim and epoetin are two biologic medicines that offer significant benefits in cancer care that are now patent-expired. Biosimilar versions of both have been approved in Europe by the EMA since 2007. Using the generic model, these should both improve access to better supportive cancer care and save on costs, and permit the established budget to be reallocated into novel areas of treatment.62–64 A number of biosimilars to filgrastim have been approved for all indications of the reference product.14 The approvals were based on data that included direct comparisons to the reference product using analytic methods to indicate similarities in molecular structure, in vitro properties, PK and PD properties, and mechanism of action. In addition, efficacy and safety studies were conducted in patients with cancer.
Impact of biosimilar filgrastim
Since 2008, several versions of biosimilar G-CSF filgrastim have been approved by the EMA to match the safety, efficacy and quality of the reference product filgrastim-Amgen (brand name Neupogen®, Amgen, Thousand Oaks, California, US) (Table 4).65,66 Filgrastim was approved as the reference drug67 on the basis of effectiveness in patients treated with chemotherapy regimens that are associated with a 40% or greater risk of febrile neutropoenia (FN).68 Since then the indications have been broadened to include primary prophylactic treatment when the risk of FN is predicted to be ≥20%, with the aim of reducing this to <10%, and in secondary prophylaxis where delays or dose reductions of chemotherapy could worsen cancer outcomes. A risk of ≥20% of FN can be predicted because either the chemotherapy regimen has sufficient dose-density to cause a rate of FN of >20%, or because the patient had a lower risk drug regimen but has individual risk factors, such as age or renal disease, predicting FN rates of >20% (summarised in Table 5).69
Biosimilar filgrastim has been proven to increase access to supportive cancer care. In the UK, it improved the quality of cancer chemotherapy in London with a five-fold increase in patient access.70,71 At the same time, it delivered budget savings for reinvestment.70,71 This is not an observation unique to the UK, as similar impact was shown in southern Sweden with again a fivefold increase in access to treatment and concomitant budget savings from biosimilars,70 and also in New Zealand where biosimilars have been reported to have transformed chemotherapy safety.72 Across Europe, all countries saw a jump in the use of filgrastim following EMA approval.73 However, not all countries have achieved savings and benefits from biosimilar filgrastim. In 2013, some countries managed only 2% biosimilar use, while others such as Norway and Sweden exceeded 80% use.74
Safety and effectiveness of biosimilar filgrastim
The safety and efficacy of biosimilar filgrastim has been predicted by regulators from the totality of evidence for the comparability of physicochemical structure, bio-potency, toxicology, PK and dynamic studies, as well as comparative clinical trials. These are confirmed by formal post-marketing (phase IV) clinical studies, real-world public databases and from the European drug vigilance data. The systematic review of randomised trials (15 studies, n=3,182) comparing all G-CSF (daily-dose filgrastim or lenograstim and long-acting peg-filgrastim), by Kuderer et al. showed that use of any G-CSF was associated with a statistically significant reduction in the number of patients with one or more episodes of FN, compared with control: 22.4% versus 39.5% (random effects risk reduction [RR]: 0.54, 95% confidence interval [CI]: 0.43, 0.67; p<0.0001). The effect was seen in all age groups, blinded and unblinded studies and for studies permitting secondary prophylaxis with G-CSF in control groups.75
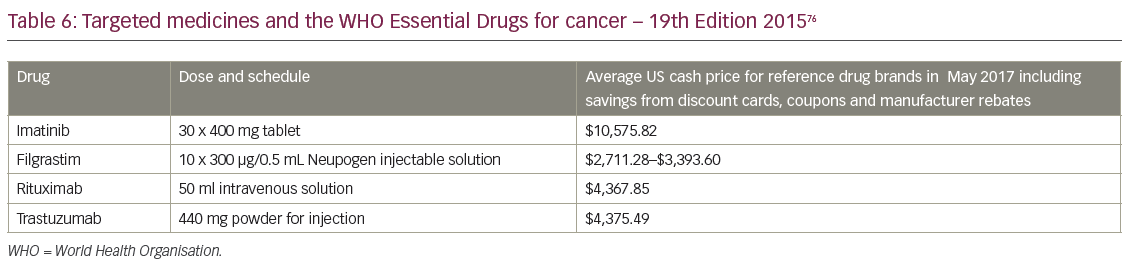
The WHO issues lists of drugs that have been deemed essential to cancer care. Medicines are included for two reasons: they deliver significant proven long-term health benefits and they are affordable. WHO member nations commit to provide such drugs at free or affordable prices to all citizens (Table 6).76 The 19th edition of Model List of Essential Medicines, published in 2015, included four targeted high-cost cancer drugs for the first time. Importantly, they listed filgrastim for supportive cancer care. These four medicines are listed in Table 6 alongside current US cash prices using discount cards, coupons and patient assistance programs, on 18 May 2017 from Drugs.Com.77–80 For many nations reimbursement of these medicines will only be possible because of the approval of biosimilar versions with assured quality, sold at a price the individual and the community can afford.

Success in the real-world setting has been shown for all the available biosimilar agents, including for example, filgrastim-Hospira (Nivestim, Hospira UK Ltd., Maidenhead, UK) in two long-term observational studies (NEXT and VENICE), and filgrastim-Sandoz (Zarxio®, Sandoz, Upper Bavaria, Germany), where five post-marketing studies have been pooled for meta-analysis.70,81 The expected outcomes from large observational studies are summarised in Table 7. Studies were selected from the database used in the systematic reviews by Pfeil et al. in 201582 and by Cooper et al. in 2011.83 This was supplemented with a search for observational studies using daily G-CSF (filgrastim or lenograstim) in the ClinicalTrials.gov database, in abstracts from the annual scientific meetings of ASCO, ESMO, ASH, EHA and MASCC, and the PubMed database.
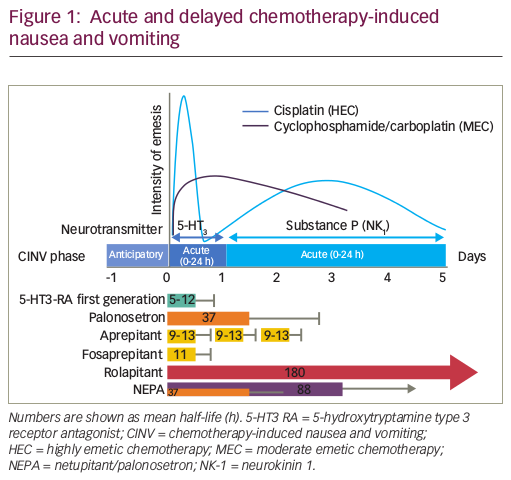
An example of a post-authorisation study is ‘NEXT’ (Tolérance de Nivestim chez les patiEnts traités par une chimiothérapie anticancéreuse cytotoXique en praTique courante: ClinicalTrials.gov Identifier: NCT01574235), which assessed biosimilar filgrastim safety given as prophylaxis in 2,012 patients undergoing cytotoxic chemotherapy for malignancies (excluding chronic myeloproliferative and myelodysplastic syndrome).81,84 It was a French multicentre, prospective, longitudinal, observational study evaluating the safety profile of filgrastim-Hospira in real-world clinical practice. The study design is summarised in Figure 1.84 Participants were aged 63.5 ± 12.7 years, 50.2% were male, 75% had a solid tumour rather than haematological malignancy and 98.2% received filgrastim for prophylaxis against chemotherapy-induced FN. Filgrastim was prescribed as primary prophylaxis in 92.6% and secondary to 7.4% of patients. Primary prophylaxis was given in 22.6% of patients because either the chemotherapy regimen was expected to cause a rate of FN of >20%, and in 77.4% because the patient had a lower risk drug regimen but had individual risk factors, such as age or weight loss, predicting FN rates of >20%.65 Filgrastim-Hospira was started 2 days after chemotherapy (median) for a mean duration of 6.0 ± 3.8 days.

Clinical outcomes assessed in NEXT were the occurrence of FN, the impact of FN, and infections on the patients and on conduct of subsequent chemotherapy cycles. Impact assessments included the proportion of patients hospitalised for FN or infection, duration of hospitalisations, the proportion of patients with reductions in chemotherapy dose, and the proportion of patients whose chemotherapy was postponed. Adverse events were also assessed. There are toxicity risks reported with the use of filgrastim. The most consistently observed toxicity is mild-to-moderate bone pain in 10–30% of patients, which is usually controlled by mild analgesics.85–87 The additional risks of nausea, fatigue, alopecia, vomiting, and headache are reported equally in placebo and treatment groups suggesting that this is probably caused by the chemotherapy itself.
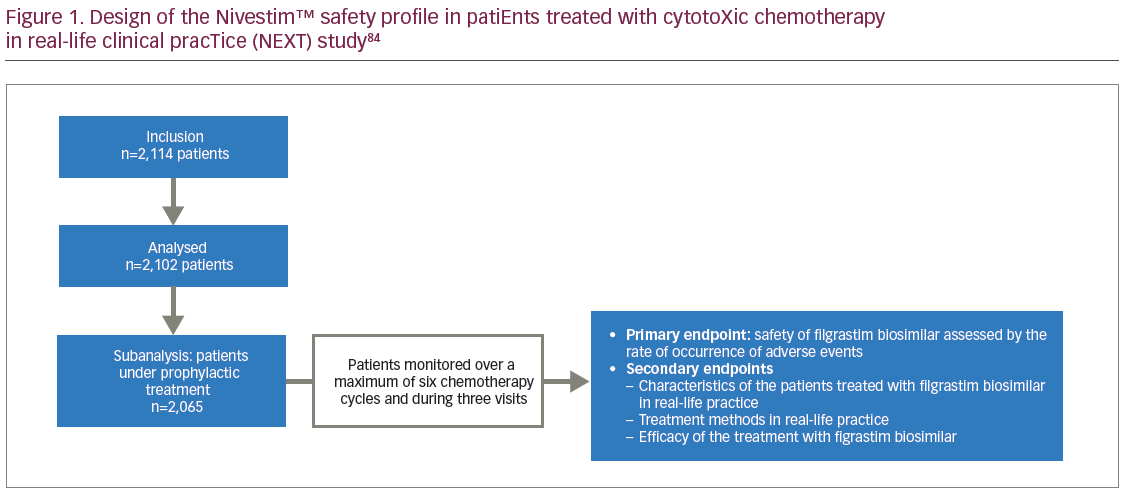
In the NEXT study, 4.9% of patients experienced FN (95% CI: 4.06–5.98), and all were hospitalised for a median of 5.5 days (mean 8.7 days). Infections were identified in 3.1%. Chemotherapy dose was reduced in only 4.7% and delayed in 7.4% of patients. Adverse events were seen in 20.4% of patients: 12.7% presented with bone, muscle, and/or chest pain, 3% nausea, 2.3% diarrhoea, and 1.8% headache.81,84 This is consistent with the outcomes expected for reference filgrastim or other daily-dose filgrastims or lenograstim in observational studies (Table 7), and from the pivotal regulatory trials of the biosimilar.88 Table 7 summarises the other large observational studies (with ≥200 patients) of daily-dose G-CSF given as prophylaxis against FN in studies reporting one or more clinical outcomes.89–97
In a further real-world observational study from Portugal, patients with dose-dense docetaxel, doxorubicin, cyclophosphamide (TAC) chemotherapy for breast cancer showed similar outcomes of 16% FN rate in 147 patients, with reference filgrastim-Amgen, and also in 134 patients with biosimilar filgrastim-Hospira.98
Observational studies are able to show that even in the most critical use of filgrastim in stem cell transplantation, biosimilars are effective. A pooled analysis of the 12 autologous and five allogeneic healthy donor biosimilar cell mobilisation studies to date showed there are no significant differences between biosimilar versus originator G-CSF in the median number of CD34+ cells mobilised (frequency in peripheral blood or dose of apheresed CD34+ cells by body weight), or in the number of G-CSF injections and leukaphoresis procedures required to harvest the target CD34+ cell dose.99
Safety of switching between reference and biosimilar filgrastim
Switching from one brand of filgrastim to another within the same patient has proved safe: no difference was recorded in 10 studies included in a meta-analysis from the Utrecht Institute for Pharmaceutical Science.100 Repeated switching back and forth between brands for each of six chemotherapy cycles has also proved safe for biosimilar filgrastim in the phase III study comparing the efficacy and safety of EP2006 and filgrastim (Pioneer) trial.101,102 Induced anti-drug immunity with biosimilar filgrastim has been absent. For example, no signs of induced immunity have been reported in patients with breast cancer and healthy volunteers treated with filgrastim-Sandoz.70
All biosimilar filgrastims approved by the EMA in 2008 have passed 5 years of use. This milestone is marked by EMA with a review of the post-marketing trial and safety data. All the approved filgrastim biosimilars have been reapproved for all indications.103 By 2015, the majority of filgrastim use in Europe was with biosimilars.104 This demonstrates the success, to date, of the EMA pathway, which has proved sufficiently robust to deliver biosimilar medicines that have matched reference outcomes, as well as achieving physician confidence.
Impact of biosimilar epoetin
Anaemia is the most common haematological complication observed in patients with cancer.105 In the European Cancer Anaemia Survey (ECAS), 67% of patients with cancer had Hb levels of <10 g/dl during a 6-month period.106 Data from the same survey highlight that anaemia worsens as chemotherapy progresses and is frequently under recognised and under treated. Of 9,118 patients with anaemia, less than 40% received any anti-anaemia treatment (erythropoietic protein, transfusion and/or iron). The 2014 UK National Institute for Health and Care Excellence (NICE) review of erythropoiesis-stimulating agent (ESA) trials for chemotherapy-induced anaemia (CIA) performed a meta-analysis to summarise the benefits and risks of this approach.107 This showed that, compared with a transfusion policy, ESAs improved Hb over chemotherapy by 1.59 g/dl (CI: 1.33–1.84). All the individual trials showed a beneficial effect of ESA treatment on Hb response with little heterogeneity (I2=6.4%; p=0.383), all but one study showed a beneficial effect of ESA treatment with less transfusions and 13 trials with quality of life endpoints confirmed the benefits of ESAs on wellbeing. When used for the labelled indication of treating anaemia during cancer chemotherapy to Hb targets of ≤12g/dl, the UK NICE review was able to show no detrimental impact on survival or tumour response. A similar result was seen in a second meta-analysis in 2014 that included only trials in patients with cancer with a baseline Hb at <11 g/dL and a target Hb ≤13 g/dL, with an improvement in anaemia, reduced red blood cell (RBC) transfusion frequency, and no increased on-study mortality or cancer progression in cancer patients with anaemia.108 This quality-of-life benefit without survival disadvantage for on-label ESA use is confirmed by a similar meta-analysis from 2009, and confirms the useful supportive care role of ESAs in cancer treatment.109
Biosimilar epoetins have been proven to increase access to supportive cancer care (Table 8)67,68,110 by reducing costs.62 No stronger demonstration could be seen than the impact in the UK. The first health technology assessment of epoetins and ESAs for supportive care in cancer by UK NICE in 2008 concluded that the drugs were clinically effective in reducing transfusions and improving the quality of life during chemotherapy, but were not cost effective at list price.111 However, a second NICE appraisal 5 years later accepted that the impact of biosimilar competition had reduced prices significantly, such that list prices were no longer appropriate to determine cost effectiveness.107 Using these real-world prices, NICE showed that epoetins for CIA had a cost quality adjusted life year (QALY) gain of less than £20,000 ($31,000; €27,000) and approved epoetins for UK reimbursement. The impacts of biosimilar epoetins may be significant, especially in the US, where for several years, epoetin alfa was the single greatest drug expenditure paid for by US Medicare.112
The naming of epoetin biosimilars in Europe can seem inconsistent.113 The reference product for all the biosimilars approved by EMA was epoetin-alfa Eprex (Janssen-Cilag, Beerse, Belgium); however, some drugs have different Epoetin INN suffixes (epoetin-theta/zeta). Although all of the biosimilars approved using epoetin-alpha as a reference product have differences in glycosylation from the reference product, they all showed no clinically meaningful differences in the comparability exercise required for registration by the EMA.
Safety and effectiveness of biosimilar epoetin
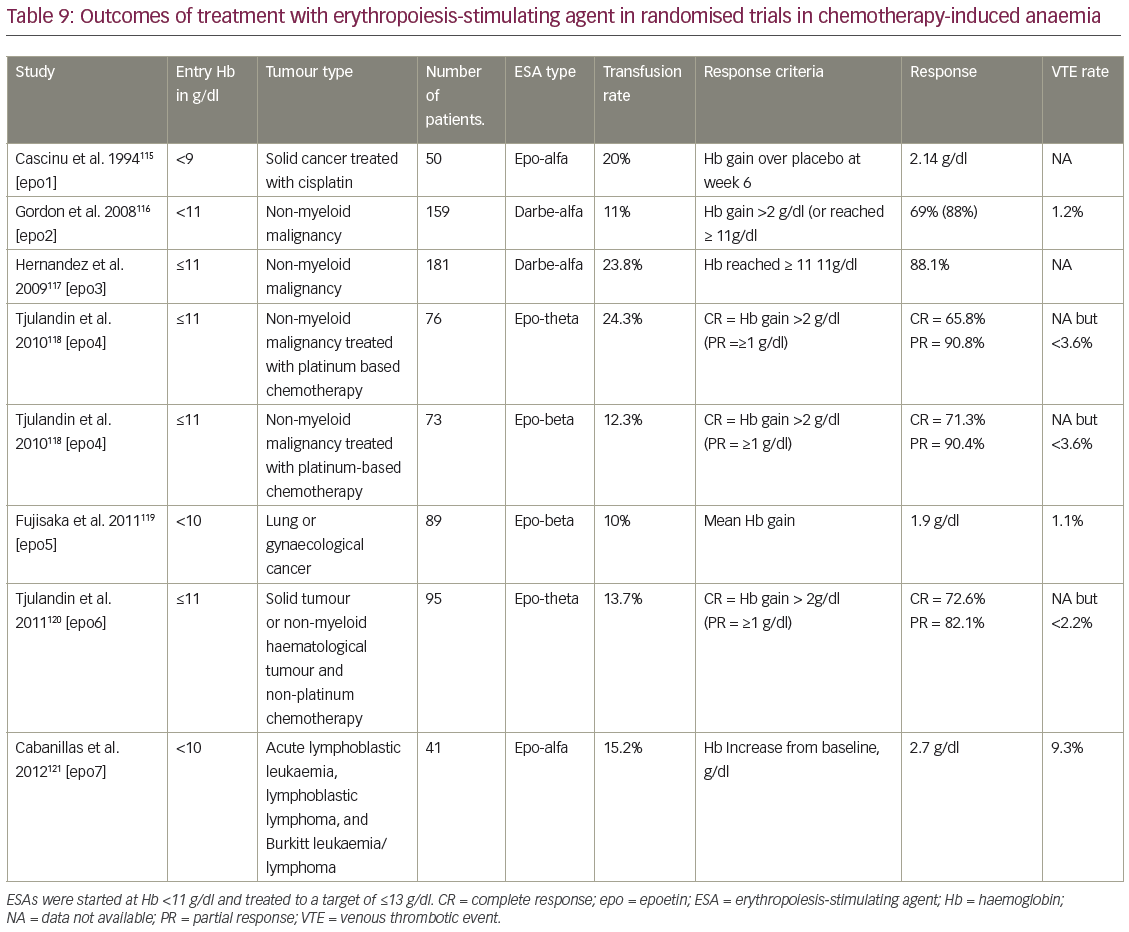
Post-approval safety of biosimilar epoetin has been assessed in a significant number of patients. For example, with erythropoietin-zeta (Retacrit®, Hospira UK Ltd., Maidenhead, UK) there have been more than 35,000,000 patient-days’ exposure, without evidence of increased complications.114 Comparison of ESA use in real-world studies has been complicated because many patients were treated with historic protocols that now differ from the current labelling indication and best guideline practice. For this reason, we have summarised data from the ESA arms of randomised trials that treated CIA with anaemic patients, according to the current EMA label in Table 9.115–121 This can be compared with the observational studies available for biosimilars and reference epoetin-alfa used according to the 2015 label summarised in Table 10.122–128 This requires studies with ESA treatment starting at Hb <11g/dl, and continued to treat to an Hb target of ≤13g/dl.108 The measures of treatment response vary between trials, but one or more measures of Hb response or transfusion rates are usually reported. Overall toxicities in chemotherapy trials will be expected to be high, but the rate of venous thrombotic events (VTE) (deep vein thrombosis and pulmonary embolus) are perhaps the most clinically important, as they may be the cause of increased mortality from ESA use in off-label indications with VTE, the second most frequent cause of death in cancer patients.129 In addition to the studies in Table 9, data from more than 2,000 patients from ESA biosimilar treatment of CIA will be published from the French prospective Biosimilar Retacrit in the Treatment of Chemotherapy-induced Anaemia in Oncology and Haematology (SYNERGY) study.130
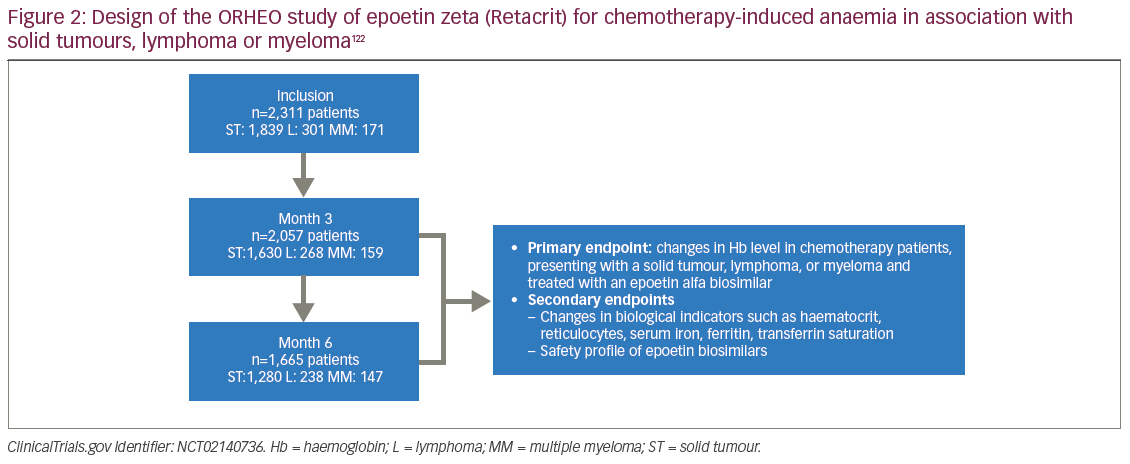
The largest of the formal post-authorisation studies for biosimilar epoetin used for CIA is the ORHEO study of epoetin zeta (Retacrit), summarised in Figure 2 (ORHEO: place of biOsimilaRs in the therapeutic management of anaemia secondary to chemotherapy in HaEmatology and Oncology).122 The ORHEO study prospectively followed 2,311 adult patients with solid tumours, lymphoma and multiple myeloma who developed anaemia during chemotherapy and were treated according to the current European Organisation for Research and Treatment of Cancer (EORTC) anaemia guidelines and 2015 epoetin alfa European label.105,131 The ORHEO study shows that comparable results can be achieved for treating CIA with either biosimilar and reference epoetins. The mean study baseline Hb was 9.61 g/dL, with 35.6% of patients having moderate anaemia (Hb 8–9.5 g/dL). Epoetin-zeta was given to 99.9% of participants. In keeping with the current drug label for epoetin-alfa, the aim was to treat established anaemia and restore Hb to the range 10–12 g/dL. An Hb response, defined as Hb >10 g/dL or rise of ≥1 g/dL, was achieved in 81.6% and 86.5% of patients at 3 and 6 months, respectively. Overall mean change in Hb level was 1.52 ± 1.61 and 1.72 ± 1.61 g/dL at 3 and 6 months, respectively. Transfusion and VTE rates were 9.4% and 2.4% at 3 months, and 5.8% and 1.5% at 6 months respectively, without any evidence of pure red cell aplasia (PRCA). This matches the expectations for the reference drug.
Comparisons of outcomes between epoetin-alfa reference and biosimilar drugs at a population level are available for one region of Italy. In the first report, comparable efficacy was shown with equivalent Hb responses and transfusion rates in both oncology and nephrology indications.132 The second study shows comparable safety with no significant differences in all-cause mortality, blood transfusion rates, cardiac events, blood dyscrasia diagnoses or hypersensitivity reactions: the composite outcome confirmed these results with an adjusted hazard ratio (HR) = 1.02; 95% CI: 0.78–1.33 for patients treated with biosimilars versus epoetin α originators.130
Safety of switching between reference and biosimilar epoetin
Switching has been common between reference epoetins and non-innovator brand epoetins in both the EU and US. This switch between molecules as different as epoetin-alfa, epoetin-beta, darbepoetin and methoxy polyethylene glycol-epoetin beta (continuous erythropoietin receptor activator or ‘CERA’) has proved safe with 22.0% and 14.4% of Italian and US patients in real-world studies of switching respectively.133,134 In the Italian study, 22% of patients who switched did so twice in an 18-month period.135 In the Prospective Immunogenicity Surveillance Registry (PRIMS), one in five patients switched epoetin brands without reported complications.136 In children’s therapy, 90% of paediatricians surveyed have switched a patient’s biologic drug to another brand.137 It is no surprise to find no evidence of a difference in safety profiles between reference and biosimilar brands of epoetin in clinical trial data or post-marketing surveillance data in the meta-analysis by Ebbers et al. in 2012.100
It has been suggested that biosimilar epoetins might result in immune reactions that increase the risk of neutralising anti-drug antibodies, compared with the use of reference epoetins.138 The role in which epoetins are most sensitive to developing immune reactions is with chronic use in haemodialysis and pre-dialysis. Patients with anti-drug antibody-mediated PRCA have not been observed in the oncology supportive care indication of CIA. Consistent with the observed safety in observational studies, more than 300 patients followed in a randomised epoetin-zeta Hospira Retacrit biosimilar switch trial, which showed that no anti-drug antibodies could be detected in either group.139 No cases of PRCA have been seen in the 1,634-patient post-marketing, post-authorization safety cohort observation of Silapo/Retacrit (epoetin-ζ) administered intravenously for the treatment of renal anaemia (PASCO) study,140 or in the 478 patients studied with epoetin-alfa Binocrit.141 This reassurance has been seen in a meta-analysis of 10 studies, all with biosimilar epoetins with adverse event rates comparable to reference drugs.142 Pharmaco-vigilance studies are equally reassuring, with no reports of neutralising antibodies or cases of antibody-mediated PRCA in more than 86,000 patient courses of epoetin-zeta (nearly 32,000 patient-courses have been in the oncology indications) estimated for 2008–2011.143,144
Over the surveillance period 18 December 2007–31 December 2011, an estimated 31,941 patient-courses were used according to the epoetin-zeta oncology indication. The total cumulative post-marketing exposure estimate (combined oncology and nephrology data, post-marketing and clinical exposure data) was approximately 86,159 patient-courses. No reports of neutralising antibodies or cases of PRCA have been received to date.144
The impact of biosimilars in future cancer care
The most important health policy objective is the improved health status of the population. In developed healthcare systems, the most common medical treatment is pharmaceutical therapy. Success of public health policies in improving the health status of the total population highly depends on the success of policies to control the cost of drugs and improve patient access.145 The WHO makes it clear that sourcing and using the lowest cost medicines is a requirement of good prescribing (Table 11).146

The WHO, in their 2010 World Health Report, More Health for the Money, wrote: ‘All countries can do something, many of them a great deal, to improve the efficiency of their health systems, thereby releasing resources that could be used to cover more people, more services and/or more of the costs’. The first priority amongst 10 suggested targets for increased cost effectiveness was to address the underuse of generics and higher than necessary prices for medicines.146
As with generic drugs, to get the best returns for a health system, biosimilars may need an active strategy from payers to promote their use.147,148 These could take the form of directives from payers, patient co-payments or gain-share to physicians. Already there are national plans for biosimilar switching in Poland, Finland, Denmark and Holland.149–151 This initiative is important to consider now, as 9 of the 10 best selling drugs in the world are set to lose patent protection in the next 5 years, with combined sales of more than $50 billion in 2011 alone.152 Predictions in 2008 suggested that even with modest price discounts of 25–35%, biosimilars could save the US between $67 and $108 billion between 2010 and 2019.153 As well as switching between reference and biosimilar drugs, there is a further potential gain to be achieved by therapeutic substitution, known also as drug switching and therapeutic interchange. This is the practice of replacing a patient’s prescription drugs with chemically different drugs that are expected to have the same clinical effect. With short-acting filgrastim and epoetin now available as less expensive biosimilars, prescribers should also consider therapeutic substitution within departmental protocols.154
At a global level, the impact of biosimilars in oncology will be significant. The WHO Model Lists of Essential Medicines for adults (EML) and children (EMLc) presents a set of medicines that are considered to be cost effective and of critical public health importance in all countries.155 The approval of biosimilar filgrastim in regions such as the EU, US and Japan, and the patent expiry and likely imminent arrival of biosimilar trastuzumab156 and rituximab157 has enabled all three drugs to be placed on the latest WHO essential drugs list.76 Critical to the inclusion in the essential drugs list is affordability, for the WHO indicates these drugs ‘should therefore be available at all times in adequate amounts and in appropriate dosage forms, at a price the community can afford.’ (Personal communication from the WHO to the Executive Board, January 2002).
Developments in therapeutic oncology biosimilars
The positive experience of comparable biosimilars in cancer supportive care suggests that WHO-compliant regulatory pathways should be able to deliver copies of other patent-expired biologic cancer medicines. Regulators are known to be assessing proposed biosimilar trastuzumab, bevacizumab and rituximab in 2017.
Trastuzumab (Herceptin®)
Herceptin® (F. Hoffmann-La Roche Ltd., Basel, Switzerland) is a humanised MAb indicated for treating human epidermal growth factor receptor 2 (HER2)-positive breast cancer and HER2-positive metastatic gastric or gastroesophageal junction adenocarcinoma.14 As this drug has a number of indications, the study population in clinical trials may influence the confidence of clinicians for extrapolation to the other indications, and the choice of endpoint is also important.158 Several biosimilars are in development, including ABP 980, BCD-022, CT-P6, MYL-14010, PF-05280014 and SB3.61 Clinical trials have been initiated with ABP 980, CT-P6, MYL-14010 and PF-05280014, and have generally showed equivalence to the originator. Three biosimilars have been approved by the Korean Ministry of Food and Drug Safety (Herzuma® Celltrion Inc., Incheon, South Korea) and the Drug Controller General of India (Hertraz™ [Mylan, Canonsburg, Philadelphia, US] and CANMAb™ [Biocon, Bangalore, India]).158 One biosimilar trastuzumab has been positively assessed with unanimous voting by the Oncology Advisory Committee of the US Food and Drug Administration (FDA), but has yet to receive marketing authorisation.159
Bevacizumab (Avastin®, Genentech, California, US) is a recombinant humanised MAb approved in the EU and US for a number of indications including in combination therapy in metastatic colorectal cancer, metastatic or recurrent non-squamous non-small-cell lung cancer and renal cancer, and cervical cancer.14 The first approved biosimilar bevacizumab was approved in the US in September 2017 as bevacizumab-Amgen.160

Rituximab (Retuxan®, MabThera®)
The MAb rituximab is approved for both oncology and anti-inflammatory indications, being a chimeric murine-human MAb directed to the CD20 antigen of B cells.14 In 2017, the EMA approved rituximab-truxima (Celltrion Inc., Incheon, South Korea), with several other proposed biosimilars currently under EMA evaluation.161
Development in biosimilars for other non-oncological disorders
The first biosimilar MAb to be approved by the EMA was infliximab (CT-P13) in 2014.14 This is a tumour necrosis factor inhibitor and its efficacy and safety was demonstrated in clinical trials of rheumatological disease. The findings were subsequently extrapolated to other indications. Systematic reviews of infliximab biosimilars confirm comparable outcomes for both directly tested and extrapolated indications.162–164 Switching studies with infliximab, such as NOR-SWITCH (the NOR-SWITCH Study, NCT02148640), further confirm that biosimilar MAbs are likely to be as comparable as earlier generation agents.165
The importance of engaging patients in the decisions on generics and biosimilars
While there is a significant public health benefit to be gained from biosimilars, there is evidence that patients have anxieties about generic and biosimilar medicines, which could be made worse by payer directives that restrict patient choice.166 Physicians and allied health professionals need to understand these concerns and learn how to respond to them. Such anxieties can translate into lower compliance with important consequences for patients with worse clinical outcomes, and emphasises why physicians need to engage patients and patient advocates over biosimilars in advance of their use.167 In the UK, NICE has presented a series of examples of current practice to guide centres through potential biosimilar switching policies.168 Similar experiences have been reported from Skane Hospital in Sweden.169
United States perspective on biosimilars
Although biosimilars have been available for clinical use in Europe for 10 years, the first US biosimilar was approved in March 2015 (filgrastim-Sandoz, Zarxio; Sandoz, Holzkirchen, Germany). Despite the fact that the FDA regulatory pathway for biosimilars is based on the successful EMA experience as well as the FDA’s experience in approving reference biologics that have undergone manufacturing changes, one survey of US physicians found that 35% would be reluctant to prescribe a biosimilar, and 85% of respondents desired veto power over biosimilar substitution.170
Significant educational efforts will be required to reassure US physicians about biosimilar safety and efficacy. The economic implications of biosimilar adoption will be enormous in the US. As noted above, 8 of the top 10 drugs on Medicare’s expenditure list are biologics. All eight of these drugs have either already lost patent protection or will lose patent protection by 2018. Biosimilars offer a practical approach to ‘bending the healthcare cost curve’.
The overwhelming approval of a first US biosimilar MAb, infliximab-CT-P13, by the FDA Arthritis Advisory Committee in Washington on 9 February 2016 shows further confidence in the US regulatory pathway for biosimilars. Signalling endorsement of the scientific basis of biosimilars, the advisory committee approved the biosimilar for all the extrapolated indications despite clear differences in non-critical drug attributes between the biosimilar and the reference drug.171 Vital to approval was the strong endorsement by the FDA itself in its briefing document to the advisory committee and in its positive public statements to the committee during the meeting.172,173
Conclusions
Biosimilars offer significant benefits for supportive care in cancer, primarily in increasing patient access to effective treatments. To date, the evidence from both regulatory and post-marketing studies show that biosimilars approved in Europe are comparable to their reference drugs in terms of safety and efficacy. In the WHO World Medicines Report of 2011, it was stated that medicine use is rational (appropriate, proper, correct) when patients receive the appropriate medicines in doses that meet their own individual requirements, for an adequate period of time and at the lowest cost both to them and the community.174 Irrational (inappropriate, improper, incorrect) use of medicines is when one or more of these conditions is not met.