The overwhelming majority of patients diagnosed with chronic phase chronic myeloid leukaemia (CP-CML) can achieve long-term survival with a good quality of life. In a recent review, it has been suggested that CML patients responsive to tyrosine kinase inhibitors (TKIs) might become a chronic disease, such as diabetes or hypertension, requiring maintenance treatment.1 If we accept this concept, TKI exposure could raise some problems, such as emerging resistance, chronic toxicity and, last but not least, financial costs.2 The initial therapy of CML is based on three available TKIs. These drugs are able to inhibit the phosphorylative activity of the proteins that are coded by the BCR-ABL fusion gene.3–6 The TKIs registered for their activity on BCR-ABL are imatinib, nilotiniband dasatinib. Another compound – bosutinib – has been recently evaluated in first–second line therapy.7 A fifth compound – ponatinib – has been tested in advanced patients.8
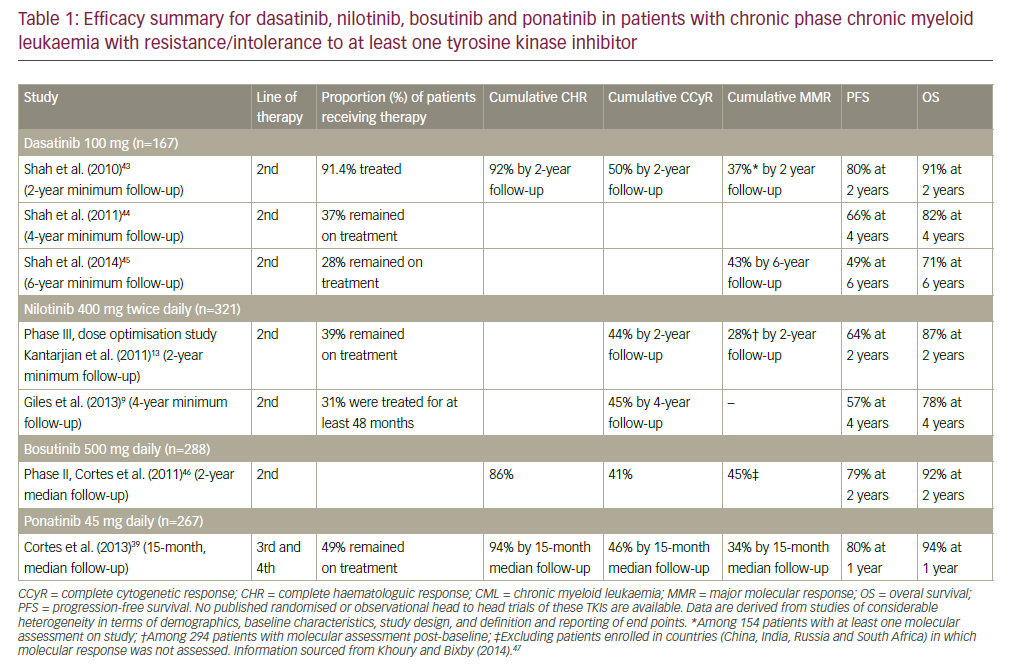
Imatinib was introduced in 1998 and changed the therapeutic landscape for patients with CML: it remains the most generallyutilised first-line therapy for newly diagnosed CML patients. A complete haematological response (CHR) is typically achieved by almost all CML patients. The complete cytogenetic remission (CCyR) rate ranges between 60–80 %, the major molecular response (MMR) ranges between 40–60 % and the complete molecular response (CMR) ranges between 15–30 %. Despite this, about one-third ofpatients who receive imatinib become resistant or discontinue the drug because of side effects.9–14 The progression-free survival (PFS) and overall survival (OS) at 10 years are in the order of 80–85 %10–13 (see Figure 1). The most important side effects of imatinib are fluid retention, myalgia and fatigue.
Nilotinib is a derivative of imatinib that was designed to target BCRABLmore specifically than imatinib. It is able to inhibit also most mutated forms with the exception of T315I mutation.15 When thisinhibitor has been used as second-line therapy, the CCyR and MMR were around 50 % and 25 %, respectively;16 when employed as firstline treatment, the achievement of CCyR and MMR were deeper, faster and superior to imatinib.17–19 The major side effects of nilotinib are elevated bilirubin, aspartate and alanine aminotransferase, lipase and amylase and peripheral arterial obliterative disease. More recently, nilotinib has been associated with an increased risk of cardiovascular events, which tend to occur later than ponatinib.19–21 These cardiovascular events (ischaemic heart disease, ischaemic cerebrovascular events and peripheral artery disease) occurred in between 6 and 12 % of patients when the drug is delivered at 300 and 400 mg BID doses, respectively.
Dasatinib is an active BCR-ABL, which was designed as a Sarc kinase inhibitor. It is able to inhibit most mutated forms with the exception of the T315I mutation. The results of CCyR and MMR are similar to nilotinib.22 When this inhibitor was employed as first-line treatment, the responses were deeper, faster and superior to imatinib.22 The major side effects of this TKI inhibitor are leuco-thrombocytopenia, pleural effusion and, more rarely, pulmonary complications. The results of bosutinib are generally similar to those of the previously described three TKI inhibitors but the differences in terms of CCyR and MMR with imatinib are less significant.7 Diarrhoea and an increase of liver enzymes are the most important side effects.
Ponatinib is a third-generation TKI able to inhibit many mutated forms, including T315I. The safety profile is not yet fully understood but when utilised after first and second-lines TKIs are able to achieve 50 % of response in highly pre-treated patients.23 Evidence has recently emerged that ponatinib was associated with a markedly increased risk of arterial occlusion.19,24–26 In the Ponatinib Ph+ ALL and CML (PACE) trial, the incidence of arterial occlusion in patients treated with 45 mg daily, was 19 % by 12 months and increased up to 29 % by 24 months.24 In order to minimise these risks, the patients should be carefully evaluated for their cardiovascular status prior to ponatinib initiation and setup of adequate prevention measures to reduce the risk.
Other available approaches for the treatment of CML are: interferon alpha (IFN-α) and allogeneic transplantation. IFN-α was considered the best drug in the pre-imatinib era when utilised at diagnosis and after autografting.27,28 Now it is utilised as pegylated preparations in combination with imatinib in different clinical trials.14 The German CMLStudy IV randomised 1,551 CML patients to imatinib 400 mg, imatinib plus IFN-α, imatinib plus arabinosylcytosine (Ara-C), imatinib after IFN-α failure or imatinib 800 mg.29,30 The best results were achieved with highdose imatinib and imatinib/IFN-α and these patients obtained molecular remission4,5 more quickly than patients on the other study arms.30
The French STI571 Prospective Randomized Trial (SPIRIT) assigned more than 600 patients to imatinib 400 mg, imatinib 600 mg, imatinib plus Ara-C or imatinib plus pegylated (Peg)-IFN-α.31 This study demonstrated at 12, 18 and 24 months significantly faster molecular response rates with imatinib plus PegIFN-α-2a compared with the other study treatments.31 Unfortunately, PegIFN-α-2a was not well tolerated, so the dose was lowered to 45 μg/week. In conclusion, these studies and others confirm that the combination imatinib/IFN-α may increase the rate of deep molecular responses but the undesirable side effects of IFN-α can create problems in delivering therapy. Allogeneic stem cell transplantation should not be considered the treatment of choice in the first-line setting. It should be indicated in patients who are in advanced phase of CML or in chronic phase but resistant to first- and second-line TKIs.
Which is the Best First-line Option?
The best first option is one of the three available TKIs. Both nilotinib and dasatinib are superior to imatinib in terms of CCyR and MMR and the number of patients who progress are lower with these two TKIs than imatinib. The results achieved in CP-CML patients treated with nilotinib showed CCyR achievement at 3 months in about 80 % of them and in more than 90 % at 6 months, whereas the MMR rates where 52 % and 62 %, respectively, and more than 80 % at 12 months.17 The phase III, randomised multicentre study (Evaluating Nilotinib Efficacy and Safety in clinical Trials [ENEST]), comparing the efficacy and safety of nilotinib versus imatinib in newly diagnosed patients demonstrated that MMR at 1 year was significantly higher for nilotinib (44 %) than for imatinib (22 %) (p<0.0001). Responses were rapidly achieved with nilotinib and were associated with less progression to advanced phases. At the last follow-up, MMR and CMR continued to be higher with nilotinib18,19 but the adverse events, in particular cardiovascular, were more frequently associated with it. In another phase III randomised multicentre study, Dasatinib versus Imatinib Study in treatment-naïve CML patients (DASISION), the efficacy and safety of dasatinib 100 mg OD was evaluated as the first-line therapy versus imatinib 400 mg/day.32 The primary endpoint of the study was CCyR at 1 year; at this time dasatinib achieved more CCyR (83 %) than imatinib (72 %) (p<0.001); also the MMR rate at 1 year was higher for dasatinib (46 %) than for imatinib (28 %). Moreover, fewer progressions to advanced phase were observed with dasatinib. After 60 months MMR and CMR continued to be higher for dasatinib (76 % versus 66 %; p=0.002). The adverse events of dasatinib were higher than those of imatinib and were prevalently pleural effusion and myelosuppression; on the contrary, vascular events were not common in the dasatinib group.
Another phase III multicentre study (Bosutinib Efficacy and safety in CNL [BELA]) recently compared bosutinib 500 mg OD versus imatinib 400 mg OD.33 The CCyR at 1 year was the primary endpoint of this study but no significant difference was found between the two drugs. It is possible that the results of bosutinib were penalised by drugrelated events and discontinuation of the bosutinib arm. Despite this, molecular remission was higher for bosutinib (39 %) than for imatinib (26 %) (p=0.002).
More recently, great importance was given to a faster time to achieve CCyR and, most importantly, BCR-ABL transcript reduction <10 % at 3 months.34–45 All authors demonstrated that an early molecular response is able to predict the improvement of overall survival.
What is our Opinion on the Best TKI Approach as Front-line Therapy?
Imatinib has changed the landscape of CML. The cost of the drug is not low and this has certainly reduced the use of this drug in many lowincome countries. In the near future, hopefully imatinib will be available as a generic (as happened already in Turkey) and this could be save more patients. On the other hand, two other TKIs, dasatinib and nilotinib, are potential alternatives to imatinib as first-line therapy for CML, mainly because of the deeper and faster molecular responses induced by these drugs.18,32 In our opinion, imatinib should be used in low-risk patients while the second-generation TKIs should be offered to intermediate- and highrisk patients only. An alternative should be to initially start with imatinib 400 mg and then to switch to a second-generation TKIs as soon as a nonoptimal response has been achieved. Of course, comorbidities and sideeffects of TKIs should be considered in the final decision. Finally, if we can evaluate the mutations at diagnosis in high-risk patients, we could be able to find mutations that could be sensible to imatinib or other TKIs; in this way, we can offer the most sensible TKI for that specific mutation. Our recent retrospective experience permitted us to find mutations at diagnosis mainly in high-but not in low-risk patients.46
The Basis of Treatment for Tomorrow
Recent progress in our understanding of CML biology has highlighted new oncogenic pathways and resistance mechanisms that are considered as highly promising targets for achieving better control of this disease and, possibly, its cure. In this context, work from Tessa Holyoake’s lab in Glasgow showed that the majority of CML progenitor (CD34+) cells undergo division in culture in the presence of growth factors.47 When TKIs were added to the culture, the proliferating cells were killed, while dormant cells that do not divide were completely refractory to the drugs.48 These dormant or quiescent cells are probably responsible for the ‘molecular persistence of the disease’, namely, the residual low level of BCR-ABL transcript positivity that is detected by quantitative polymerase chain reaction (PCR) in many cases. Since the available TKIs seem not to be able to eliminate dormant cells, the question is how to devise alternative biological strategies to eradicate these cells. Recent studies from different groups have provided encouragingpotential therapeutic approaches, recently discussed in two recent CML workshops at the American Society of Hematology (ASH).49,50 As discussed by Perrotti et al., protein phosphatase 2A (PP2A) is a tumour suppressor whose activity is inhibited in Philadelphia (Ph)-positive leukaemias, but not in normal haematopoietic stem/progenitor cells.51,52 Fingolimod (FTY720), an immunosuppressive synthetic sphingosine analogue,53 has the capacity to re-activate PP2A and shows limited toxicity on normal cells. FTY720 indeed has anti-leukaemic activity on CD34+ cells from TKI-sensitive and TKI-resistant CML progenitors.54–57 Notably, in primitive CML cells, the pro-apoptotic effect of FTY720 might not require BCR-ABL activity,53 which, as reported, is dispensable for their survival.58 Since FTY720 is active on oncogene-driven pathways other than the one controlled by BCR-ABL and preserves normal cells at the same time, it is not surprising that this is also effective in other types of leukaemia.
Another interesting approach for treating CML is the inhibition of autophagy. CML cells that survive TKI treatment show features that are reminiscent of those observed in response to growth factor deprivation, such as a reduced size and a marked increase in cytoplasmic vacuoles.59 These cellular adaptations reflect the activation of autophagy, a cellrecycling process that relies on a specific self-digestion apparatus and confers the ability to survive stress conditions and inhospitable environments.60 A classic marker of autophagy-activation is represented by the cleavage and lipid modification of the Atg protein LC3, which, as a result of these modifications, is able to bind to the internal membrane of autophagic vacuoles and to act as a docking site for proteins to be ‘digested’. Treatment of CML stem cells with TKIs induces LC3 conversion and activates the autophagic programme, thus contributing to the CML stem cell resistance of these agents.61 These observations provided the rationale for combining TKIs, which kill only mature BCR-ABL+ cells, with autophagy inhibitors, such as chloroquine or hydroxychloroquine, to eradicate the CML stem cell compartment.62 A clinical trial based on these interesting results is ongoing in the UK; patients who achieve a MMR after 1 year of imatinib treatment are randomised to continue imatinib alone or imatinib plus chloroquine. Follow-up data are not available yet.
Recent laboratory studies with macrolides, such as clarithromycin, have demonstrated that these antibiotics enhance the activity of TKIs in CML cells. The mechanism of induction of cell death by clarithromycin combined with TKIs appears to rely on the inhibition of the late stage of autophagy. This is clearly demonstrated by an increase in LC3-II protein levels and by a concomitant increase in cellular vacuole formation.63 Remarkable responses obtained in seven patients with advanced CML who were treated in Genoa with clarithromycin plus TKIs indeed support the hypothesis that autophagy inhibition may make CML cells sensitive to killing by TKIs.64,65
The oncogenic Hedgehog (sHH) signalling plays a crucial role in cancer because of its involvement in chromatin remodelling, cell cycle control and apoptosis.66 The expression of the sHH effectors Gli-1 and, in particular, Gli-2, is significantly increased in both chronic and accelerated phase CML progenitors.67,68
Finally, drugs with the ability to block BCR-ABL mutated forms that are otherwise resistant to traditional TKIs are also a new frontier in the CML therapeutics arena, and some of these agents have already reached advanced clinical evaluation phases. The T315I BCR-ABL-mutant protein is resistant to imatinib, dasatinib, nilotinib and bosutinib, and remains a major concern for clinicians. Among the new TKIs, ponatinib is active against this type of mutated BCR-ABL. Ponatinib, in particular, is considered a pan-BCR-ABL inhibitor, which potently inhibits the T315I BCR-ABL mutant and overcomes mutation-based resistance.69,70
Discontinuation of TKI Therapy – Is it Possible?
Long-term TKI treatment leads to a gradual reduction of residual disease in the majority of patients and in some patients no residual disease is detectable with real-time quantitative (RQ)-PCR. The question now is: can TKI be discontinued in these particular group of patients, since such a possibility would have a major impact on patient quality of life and, in terms of costs, on healthcare systems? Recently a variety of clinical studies have evaluated the possibility of exploring the TKI discontinuation in patients with stable molecular remission. After thefirst positive results achieved with imatinib discontinuation,71 a pilot multicentre study entitled Stop imatinib (STIM) was organised; 100 patients with CP-CML and undetectable BCR-ABL transcript for at least 2 years after imatinib were followed. After 1 year, 41 % of patients were molecular relapse-free survivors.72 A recent update at 5 years confirmed a cumulative incidence of 61 % of molecular relapses with three patients relapsed at 19, 20 and 22 months.73 All molecular relapsed patients were responsive to re-treatment with imatinib. Similar results were achieved with Australia74 and Japan75 teams. In the attempt to define prognostic markers to increase the rate of patients in molecular response after TKIs discontinuation, the preliminary results of the European Stop Tyrosine Kinase Inhibitor (EURO-SKI) study have been presented at the last ASH meeting. Seven hundred patients entered the trial. An interim analysis of the first 200 patients from eight European countries with a minimum follow-up of 12 months was presented. One hundred and eleven (56 %) of the 200 patients remained therapy-free after the discontinuation of TKIs and 89 relapsed at a molecular level. Similar to the STIM study, most relapses occurred quickly within the first 6 months. The duration of prior TKI treatment and the achievement of MR4 for more than 5 yearsbefore discontinuation seemed to have a positive effect. Moreover, the estimated drug-related savings to healthcare systems in the eight EURO-SKI countries was estimated to be €7 million.
The STOP-2G-TKI study, conducted by the French CML study group, recruited 52 patients.76 The inclusion criteria of this study required prior second-generation TKI treatment with achievement of MR4.5 remission for at least 2 years. Fifty-two patients with a median of 6.5 years of TKI treatment and a median duration of MR4.5 of 2.3 years, have been evaluated and followed for 12–60 months. Twenty-four (46 %) of the 52 patients experienced molecular relapse and had to restart treatment. One relapse occurred 24 months and one 37 months after TKI discontinuation.
In conclusion, according to these results, it seems obvious that patients need to be regularly monitored even years after stopping treatment. The good news is that the molecular relapsed patient, after restarting treatment, achieved a deep molecular response and, until now, none of the patients progressed to advanced CML phases. It may be concluded that molecular response and imatinib treatment were the most important predictors for successful therapy. Recently, of great interest, are two abstracts presented at the last ASH meeting. The first demonstrated that age <45 years combined to detection of BCR-ABL in a more sensitive ‘digital PCR’ test were associated with an increasing risk of relapse.77 The second abstract (Korean Imatinib Discontinuation Study [KIDS]) confirmed that both duration of imatinib treatment and of deep molecular response were the most important predictors for successful discontinuation.78
Conclusions
Recent progress in CML stem cell biology has identified several main molecules and signalling pathways that are involved in the maintenance of CML stem cells, offering possibilities for exploring new therapies for this disease. However, much work remains to be carried out – work that should benefit new CML patients in the future and, hopefully, also patients with other forms of cancer whose management may be based on principles derived from pioneering work in CML.