Although the outcome of patients with acute myeloid leukaemia (AML) has improved with cytarabine and anthracycline-based chemotherapy regimens in addition to advances in supportive care, relapse remains frequent and constitutes the leading cause of mortality.
Although the outcome of patients with acute myeloid leukaemia (AML) has improved with cytarabine and anthracycline-based chemotherapy regimens in addition to advances in supportive care, relapse remains frequent and constitutes the leading cause of mortality.
In a study of 243 patients with relapsed/refractory AML by Keating et al., 33% patients obtained a complete remission (CR), 24% died prior to achieving a response and 43% were resistant to their first salvage regimen.1 The median survival was 18 weeks and five-year survival was only 5%. Whereas prior therapy with cytarabine or stem cell transplant (SCT) did not influence prognosis, duration of first remission was significantly associated with subsequent outcome.
In a recent article, Breems et al. defined a prognostic score for patients with AML in first relapse based on the following variables: relapse-free interval from first CR, cytogenetics at diagnosis, age at first relapse and SCT before first relapse.2 Three prognostic subgroups were identified. Eighty-five per cent achieved CR in the favourable group compared with 60 and 34% in the intermediate-and poor-risk groups, respectively. The five-year survival rates were 46, 18 and 4% in these three groups. The prognostic impact of these variables was independent of salvage therapy, and hence was most likely attributed to intrinsic disease (e.g. cytogenetics) and host characteristics. Giles et al. reported results of 594 patients who underwent second salvage therapy for relapsed AML.3 Thirteen per cent achieved CR (median CR duration: seven months), and one-year survival was 8%. Multivariate analysis revealed the following variables as independent adverse prognostic factors: first CR duration more than six months, second CR duration more than six months, first salvage therapy not including SCT, non-inversion 16 AML, platelet count <50x109/l and white blood cell (WBC) count >50×109/l. Duration of first remission was the most important predictor of response to salvage therapy.
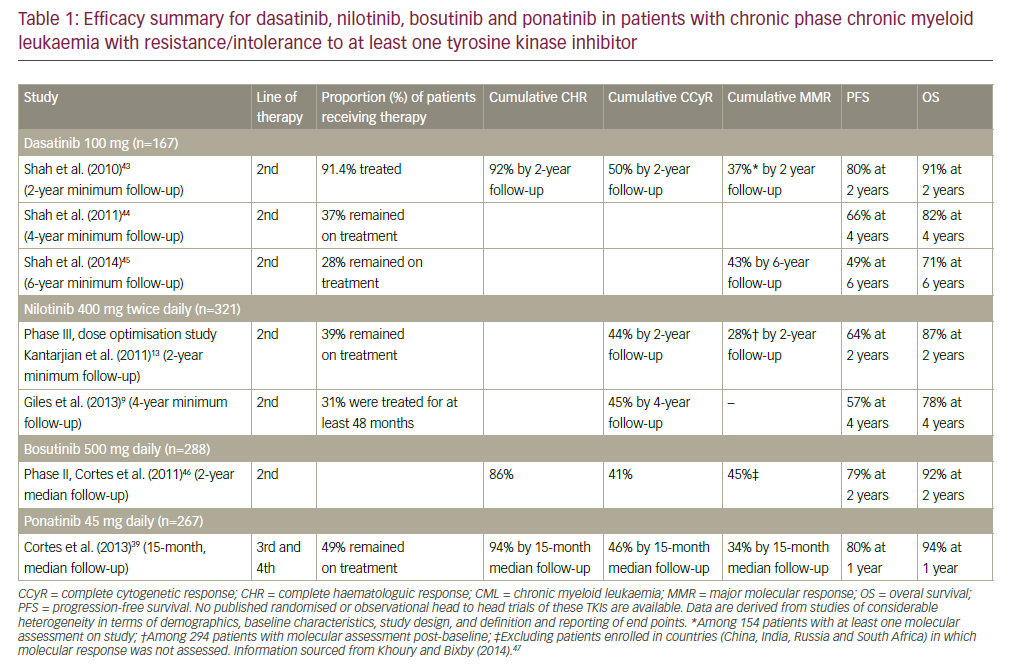
Conventional chemotherapy for relapsed/refractory AML has largely been unsatisfactory and most patients do not achieve long-term durable remissions. The only potentially curative option remains SCT, but effective regimens are necessary to control AML relapse prior to transplant. With the advent of molecular studies and better insights into the pathogenesis of AML, more specific, mechanism-driven, targeted drugs are being developed. The following sections describe a number of new drugs that have been tested in clinical trials for patients with relapsed/refractory AML (see Table 1).
Gemtuzumab Ozogamicin
Gemtuzumab ozogamicin (GO) is an anti-CD33 monoclonal antibody bound to its cytotoxic conjugate calicheamicin.4 Following attachment to the surface of myeloid blasts, it becomes rapidly internalised and releases calicheamicin. In an open-label, single-arm phase II study involving 277 patients with relapsed AML, GO was administered at 9mg/m2 as a two-hour intravenous (IV) infusion in two doses separated by two weeks.5 Thirty-five patients (13%) achieved CR and another 13% achieved CR with incomplete platelet recovery (CRp <100x109/l). All patients developed grade 3 or 4 neutropenia and thrombocytopenia. Grade 3–4 hyperbilirubinaemia was noted in 29% of patients and 0.9% of patients who did not undergo SCT developed sinusoidal obstructive syndrome (SOS). Based on these data, the US Food and Drug Administration (FDA) approved the use of GO for patients with relapsed AML who are >60 years of age and who are considered unfit for conventional cytotoxic therapy.6
A number of trials exploring the combination of GO with other chemotherapeutic regimens in relapsed AML have been published (see Table 2). The most notable among these results is a study by Chevallier et al. that reported a CR rate of 50% and CRp rate of 13%, leading to an overall response (OR) of 63% in patients treated with a combination of IV GO 9mg/m2 over two hours on day four, cytarabine 1g/m2 every 12 hours IV over two hours on days one to five and mitoxantrone (MIDAM) 12mg/m2/day IV over 30 minutes on days one to three.11 Grade 3–4 neutropenia and thrombocytopenia occurred in all patients and 55% patients had documented bacterial sepsis. Grade 3–4 hyperbilirubinaemia was observed in 16% patients and SOS in 3%.
Whereas single-agent therapy with GO appears to have limited activity in relapsed/refractory AML, combinations with chemotherapy are more promising and are being actively investigated. Caution should be exercised when GO is used with other hepatotoxic agents and within three to four months of SCT to avoid the development of SOS. Reduction of doses (3–6mg/m2) in combination therapies has proved safer.
FMS-like Tyrosine Kinase Inhibitors
Mutations of the FMS-like tyrosine kinase (FLT3) gene cause autophosporylation of the FLT3 receptor tyrosine kinase, resulting in cell proliferation and inhibition of apoptosis. In particular, internal tandem duplications (ITD) of FLT3 confer an adverse prognosis and are seen in 25–35% of patients with predominantly diploid AML. Of the >20 molecules with inhibitory activity against FLT3,12 the most widely studied agents in AML include midostaurin (PKC412), sorafenib (BAY43-9006), sunitinib (SU11248), lestaurtinib (CEP701) and tandutinib (MLN518) (see Table 3).
A phase I study testing the efficacy of sorafenib in 21 patients with relapsed/refractory AML reported a >50% reduction of the peripheral blast count in 11 patients.16 Lestaurtinib was evaluated in a phase I–II clinical trial of 14 patients with relapsed/refractory AML.13 Five patients had measurable responses, including reductions in bone marrow and peripheral blood blasts. A phase II trial testing the efficacy of midostaurin in 20 patients with relapsed/refractory AML demonstrated a >50% reduction in peripheral blasts in 14 patients.14 Twelve out of 40 patients showed a >50% reduction in peripheral blasts in a phase I trial of tandutinib.15 Another novel FLT3 inhibitor, AC220, has been shown to produce objective clinical responses, including six patients achieving CR, with an OR rate of 29% out of 58 patients with relapsed/refractory AML.18 For all these agents, responses have generally been better in those patients with the mutant FLT3 compared with those patients with the wild-type FLT3. Nausea, vomiting, diarrhoea and fatigue were the most common side effects.
Unfortunately, none of these agents when used as monotherapy has led to sustained responses. Therefore, trials are under way to combine these agents with conventional chemotherapy agents to improve response rates and durability of responses. However, a phase III study of lestaurtinib with either mitoxantrone, etoposide, cytarabine (MEC) or high-dose cytarabine in relapsed FLT3 mutant AML showed similar rates of CR and overall survival (OS) in both groups.19 Combination therapies of other FLT3 inhibitors with chemotherapy are ongoing. In vitro data also suggest synergism between these agents and histone deacetylase and mammalian target of rapamycin (mTOR) inhibitors.20,21 Clinical trials combining these agents have not yet been carried out.
Epigenetic Therapy
In contrast to altering the sequence of base pairs in genes, epigenetic modifications affect gene expression by two mechanisms: DNA hypermethylation and histone modification. Both alter gene expression via architectural remodelling of chromatin and are pharmacologically reversible. DNA-demethylating agents and histone deacetylase inhibitors (HDACis) induce re-expression of tumour suppressor and proapoptotic genes, and are now widely used in patients with myelodysplastic syndrome (MDS). Recent reports also suggest that they may be of therapeutic benefit for patients with AML.
Decitabine and azacitidine are hypomethylating agents approved by the FDA for clinical use in patients with MDS. Some of these patients with a French–American–British (FAB) diagnosis of refractory anaemia with excess blasts in transformation (RAEB-T) were reclassified as AML based on World Health Organization (WHO) criteria and were retrospectively analysed separately. In a final analysis of three sequential cancer and leukaemia group B trials (n=309) with azacitidine (75mg/m2/day IV or subcutaneous [SC] injection x seven days, repeated every 28 days), 35–48% of patients in the azacitidine group with WHO AML (n=103) experienced CR, partial remission (PR) or haematological improvement.22 Among the 33 WHO AML responders in the three studies, the median duration of response was 7.3 months (range 2.2–25.9 months). Median survival time for the 27 WHO AML patients in the azacitidine group was 19.3 months compared with 12.9 months for the 25 WHO AML patients randomly assigned to observation. In a more recent study comparing azacitidine with conventional care regimens (CCR), survival at two years was 50% for patients in the azacitidine arm versus 16% in the CCR arm (p=0.0007), with similar CR rates.23
A randomised phase III study compared decitabine (15mg/m2 IV every eight hours for three days) with best supportive care in 170 patients with MDS.24 The OR rate was 17% in decitabine-treated patients compared with 0% in the placebo group. Reponses lasted for a median of 10.3 months and were associated with transfusion independence. In a more recent study, decitabine was administered at a dose of 20mg/m2 IV daily for five consecutive days on an outpatient basis to 99 patients with MDS.25 The OR rate was 32% and hence comparable to the FDA-approved inpatient schedule.
HDACis have also been studied in patients with relapsed/refractory AML. In a dose-escalation phase I study, vorinostat (suberoyanilide hydroxamic acid [SAHA]) was administered to 31 patients.26 The OR was 29% (3% CR, 10% CRp and 16% complete marrow responses). The major side effects were nausea, vomiting, diarrhoea and headache that resolved on cessation of therapy. Due to the poor response of single-agent HDACi in patients with AML, combination regimens with hypomethylating agents have been evaluated. Garcia-Manero et al. reported the results of a phase I–II clinical study combining decitabine (15mg/m2 IV daily for 10 days) with valproic acid (50mg/kg daily).27 Forty-four patients had relapsed AML. The CR rate of these patients was 14%. Another study using a combination of azacitidine (75mg/m2 IV daily), valproic acid (50mg/kg orally daily) and all-trans retinoic acid (45mg/m2 orally) in 53 patients (of whom 20 had relapsed AML) showed a CR of 33% (5% in relapsed patients).28
Nucleoside Analogues
Nucleoside analogues comprise a large group of active anticancer agents with a long history of use in haematological malignancies, including AML. Cladribine and fludarabine are two well-known drugs of this class. Cladribine (5mg/m2 IV two-hour infusion daily for five days) was studied with cytarabine (2g/m2/d IV four-hour infusion for five days), mitoxantrone (10mg/m2/d IV for three days) and granulocyte-colony stimulating factor (G-CSF) (300μg/d SC for six days) (CLAG-M) as salvage therapy in 118 patients with relapsed/refractory AML.29 CR was achieved in 21 (49%) patients, 20 (47%) were refractory, two (5%) died early and one-year survival was 43%.
Clofarabine, a second-generation nucleoside analogue, was developed to expand the antileukaemia activity of purine analogues and reduce the associated toxicity of earlier agents (such as central nervous system [CNS] demyelination associated with fludarabine).30 Through its inhibition of ribonucleotide reductase, clofarabine increases accumulation of cytarabine triphosphate in leukaemic blasts, thus providing a rationale for its combination with cytarabine. It was approved by the FDA for the treatment of relapsed paediatric acute lymphocytic leukaemia in 2004.31 In adults, clinical development has focused mostly on AML (see Table 4). Clofarabine/cytarabine combinations have produced CR rates of 24–38% in the relapsed setting. A multicentre phase III trial is currently ongoing in which patients >55 years of age with primary refractory AML or AML in first relapse are randomised to receive either clofarabine (40mg/m2 IV x five days) and cytarabine (1g/m2 IV x five days) or placebo and cytarabine. Combination of clofarabine with anthracyclines has been reported in 44 patients with relapsed/refractory AML.37 CR rates varied from 13 to 48%, but were heavily dependent on patient selection. Dose-limiting toxicities were hyperbilirubinaemia and hepatic transaminase elevations for clofarabine/idarubicin-treated patients, in addition to mucositis and diarrhoea for clofarabine/idarubicin/ cytarabine-treated patients. A phase II clinical study comparing the efficacy of various clofarabine combinations with idarubicin ± cytarabine is currently under way.
Other Investigational Agents
Many other agents are being tested in clinical trials including farnesyltransferase inhibitors (e.g. tipifarnib),38 bcl-2 antagonists (e.g. oblimersen), aurora kinase inhibitors (ZM447439 and VX-680), kinesin spindle protein (KSP) inhibitors (ARRY520 and AZD4788), X-linked inhibitor of apoptosis protein antisense oligonucleotide (AEG35156), MEK pathway inhibitors and mTOR inhibitors (temsirolimus and everolimus).
Stem Cell Transplantation
Currently, allogeneic SCT remains the only potentially curative option for patients with relapsed/refractory AML. However, patient selection bias, cross-over from chemotherapy to SCT arms, pre-treatment prognostic factors and treatment-associated morbidity and mortality make it difficult to interpret data comparing SCT with consolidation chemotherapy. Nevertheless, current data support the use of SCT in patients with primary refractory and relapsed AML.
In patients with refractory AML, outcome following allogeneic SCT is better in patients who have no peripheral blasts and <30% blasts in bone marrow prior to conditioning.39 In a study of 21 patients who were refractory to induction chemotherapy, matched sibling SCT led to a cumulative disease-free survival (DFS) of 43% at 10 years.40 In another study, Biggs et al. reported a three-year leukaemia-free survival (LFS) of 21% and a three-year treatment-related mortality of 44% in 88 patients with primary refractory AML who underwent matched-sibling SCT.41
In 126 patients with untreated first relapsed AML who underwent allogeneic SCT, Clift et al. demonstrated a five-year DFS of 28% and a cure rate of 25%.42 However, a more common approach in the relapsed setting is to induce second CR with salvage chemotherapy prior to proceeding with allogeneic SCT. The best regimen in terms of chemotherapy for this indication has not been established. While fludarabine, cytarabine, G-CSF (FLAG) ± idarubicin salvage chemotherapy followed by either matched sibling or unrelated donor SCT has shown some activity,43 other reports have shown better results with the use of 131I-labeled anti CD-45 antibody.44 In a study of 485 patients with AML in first relapse and second remission, 241 patients underwent matched sibling SCT and 244 underwent salvage chemotherapy.45 Three-year LFS was 17% in the SCT group versus 26% in the chemotherapy group (p = not significant). However, significantly higher LFS was observed in the transplant group versus the chemotherapy group in patients ≤30 years of age with CR duration greater than one year (41 and 17%, respectively), and in patients >30 years of age with CR duration less than one year (18 and 7%, respectively). Three-year treatment-related mortality was 56 and 7% with transplant and chemotherapy, respectively.
The role of autologous SCT and non-myeloablative allogeneic SCT in the treatment of relapsed/refractory AML remains under investigation. Although results from the European Group for Blood and Marrow Transplantation showed DFS of 30–35% in patients who underwent autologous SCT in second CR,46,47 these rates are not substantially different from those of patients who undergo salvage chemotherapy.48
Conclusion
Relapsed/refractory AML continues to be a therapeutic challenge. Conventional therapies are unsatisfactory due to low response rates and a lack of translation of responses into long-term disease control. A number of investigational agents targeting cytogenetic–molecular aberrations are being actively pursued. Combination trials with agents that have different mechanisms of action are currently under way. The exact sequence of therapies, combination of agents and the role of SCT in the management of relapsed/refractory AML in terms of novel therapies continue to evolve. ■