Chronic myeloid leukemia (CML) is the first malignancy to be associated with a specific and consistent chromosomal abnormality. Present in >90% of CML patients, the Philadelphia (Ph) chromosome is the product of a t(9;22)(q34;q11) reciprocal translocation, leading to a fusion between the ABL proto-oncogene to the BCR breakpoint cluster region. The deregulated Abl tyrosine kinase (TK) activity resulting from the BCRABL fusion gene is defined in the pathogenicity of CML. Under physiological conditions, Abl TK activity is tightly regulated, as is necessary owing to its role in regulating the cell cycle and associated cellular functions. However, the constitutively active TK activity of the fusion protein results in its autophosphorylation, leading to the unrestricted stimulation of molecules and signaling proteins known to function in the RAS/MAPK or Jak/Stat pathways. Such detrimental modifications to the formerly tightly regulated cellular mechanisms cause unregulated cell proliferation. Integrin-mediated cellular adhesion is also negatively affected such that myeloid precursors to bone marrow stroma are prematurely released into the circulation; this, along with decreased apoptosis of the myeloid precursors, is characteristic of the myeloproliferative state in chronic-phase CML. Because a substantial part of the malignant transformation stems from the constitutive TK activity of the Bcr-Abl fusion protein, TK inhibition has been the goal of many molecularly targeted therapies.
Current Therapeutics
Allogeneic stem cell transplantation has demonstrated curative ability, with the potential to cure 70% of patients in chronic-phase CML.
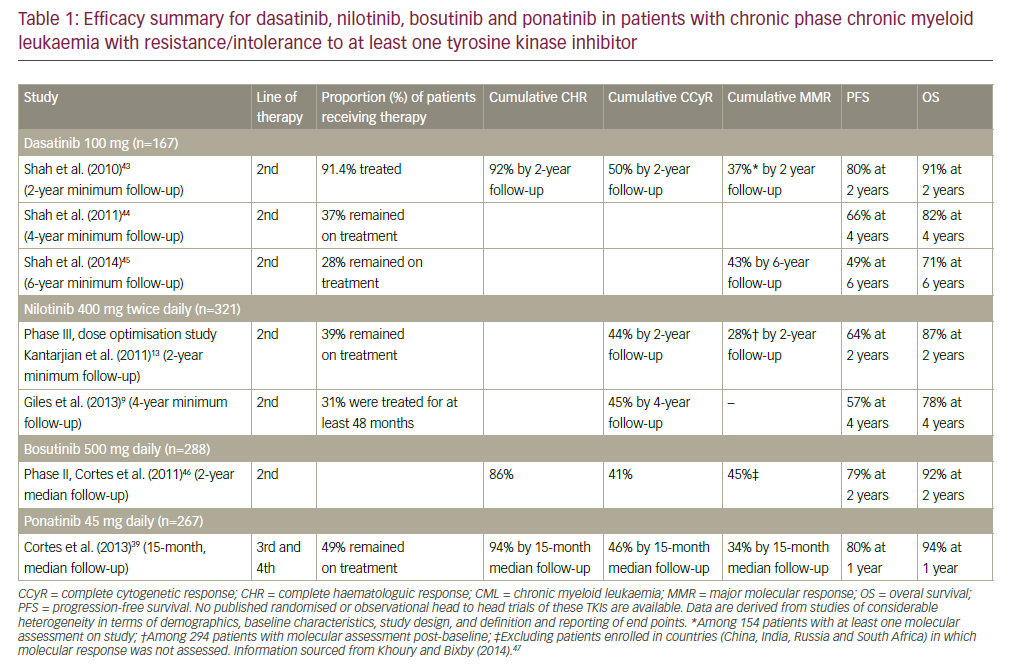
However, only 5% of newly diagnosed CML patients can actually be cured with this therapy due to limitations in donor availability, recipient age, and transplantation-associated risks.1 Interferon-α (IFN-α) has also proved to be effective, allowing 10–20% of chronic-phase CML patients to experience complete cytogenetic remission.1 After over 20 years of IFN-α as the accepted medical therapy for CML patients, this standard was overturned when the International Randomized Study of Interferon Versus STI571 (IRIS) phase III trial showed the TK inhibitor (TKI) imatinib mesylate (Gleevec, Novartis) to have superior efficacy. Almost all CML patients treated with imatinib achieved a complete hematological response (CHR), with 80–90% achieving a complete cytogenetic response.2,3 With such improvements in patient results, imatinib was adopted as the standard first-line treatment for CML in 2003. A continued six-year follow-up of the IRIS study has demonstrated obvious survival benefits with long-term use of imatinib, with treatments providing a six-year survival rate of 88%.4
Imatinib’s efficacy is due to the inhibition of the Bcr-Abl protein, with the drug occupying the TK domain and effectively negating the influence of the fusion protein on the cell cycle. In spite of imatinib’s great success, resistance is a common problem, especially in the accelerated and blast crisis phases of CML.5 Primary hematological resistance is rare (2–4% of all resistance cases) and poorly understood. Primary cytogenetic resistance is slightly more common, with 15–25% of patients failing to exhibit the desired effect within six to eight months of starting imatinib therapy.6 Another 4% of patients are estimated to develop secondary resistance each year;6 many of these cases have been associated with either acquired escape mutations in the Bcr-Abl kinase domain, allowing the protein to evade imatinib binding, or genomic amplification and continued overexpression of BCR-ABL.7–11
Although imatinib is not a curative therapy, it has been proved to prolong survival in patients with CML. Dasatinib (Sprycel, Bristol-Myers Squibb) and nilotinib (Tasigna, Novartis) have recently been approved by the US Food and Drug Administration (FDA) for the treatment of imatinibresistant or -intolerant CML patients; although active against most imatinib-resistant ABL mutants with demonstrated minimal crossresistance, there are still documented reports of resistance against these second-generation ABL kinase inhibitors.12 These drug resistances to imatinib and its successors pose significant barriers in treatment and emphasize the need for novel therapies.
Acetylation Status and Cancer
In response to the demand for novel therapies, much drug development has focused on epigenetic targets. Among these are the post-translational modifications of nucleosomal histones that control the activation or deactivation of chromatin transcription. The acetylation status of histones is determined by the co-operative enzymatic activity of the histone acetyltransferase (HAT) and histone deacetylase (HDAC) families. Acetylation can effectively modify chromatin topology and the accessibility of transcription machinery to DNA, thereby modifying the degree of gene transcription. HATs and HDACs can also modify the acetylation of lysine residues in transcription factors.13 This allows HATs and HDACs, by recruitment to promoter sites by transcriptional protein complexes, to regulate gene expression in a mechanism separate from that of chromatin remodeling. Some HDACs are also known to deacetylate non-histone, non-transcription factor proteins; among these are the nuclear import protein importin-α7,14 signal transduction protein β-catenin,15 the cytoskeletal protein α-tubulin,16 and Ku70 and WRN, enzymes involved in DNA repair.17,18 More recently, HDAC has been found to deacetylate heat shock protein 90 (hsp90).19,20 It is therefore possible that HDACs may not affect gene transcription exclusively, potentially also modifying proteins involved in important cellular pathways.
Aberrant HDAC-dependent transcriptional repression has been implicated as a main pathogenic mechanism in several cancers, including lymphoma and forms of myeloid leukemias.21,22 The BCL6 transcriptional repressor functions through recruitment of HDAC and nuclear receptor co-repressors, but in non-Hodgkin’s lymphoma lack of repression and overexpression of BCL6 result in lymphoid oncogenic transformation.23 In the M2 subtype of acute myeloid leukemia (AML), the AML1-ETO fusion protein is formed as a result of a t(8;21)(q22;22) translocation. Where AML1 will typically activate gene expression to induce differentiation in a protein complex, the recruitment of HDAC to the protein complex by AML1-ETO causes the fusion protein to act as a potent transcriptional repressor.24 The PML-RAR-α fusion protein is an oncoprotein associated with a chromosomal translocation t(15;17)(q22;21) in acute promyelocytic leukemia (APL), a subtype of AML. PML-RAR-α associates with the NCor-mSin3-HDAC transcriptional co-repressor complex to repress gene transcription and block the maturation in the myeloid line.25 In each of these examples, recruitment of HDACs appears to mediate transcriptional deregulation, providing the rationale for treatment of hematological malignancies by inhibition of HDAC activity.
Cellular Mechanisms of Histone Deacetylase Inhibitors
Inhibition in Cell Cycle and Growth
In vitro, HDAC inhibitors (HDIs) cause cell-cycle arrest in G1 or G2, as well as differentiation and/or apoptosis in cultured transformed cells. Inhibition of growth has been documented in various cell types in both hematological and epithelial cell lines. Several HDIs, including trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA), can induce the expression of the cyclin-dependent kinase inhibitor p21.26,27 This blocks cyclin-dependent kinase activity and inhibits cell-cycle progession, ultimately arresting the cell cycle in G1. Studies using complementary DNA (cDNA) microarrays show that when transformed cell cultures were treated with TSA or SAHA, only ~2% of expressed genes exhibited a change in transcription patterns.28 In addition to the CDKN1A gene, which codes for p21, HDI treatment induced the expression of genes coding for p16 (CDKN2A), p27, cyclin E, and thioredoxin-binding protein 2 (TBP2), all of which are known to regulate cell-cycle proliferation.26,27,29
Treatment with TSA can also promote telomerase activity by inducing transcription of the telomerase catalytic subunit TERT.30. SAHA and TSA have both been associated with downregulation of the DNA synthesis enzymes CTP synthetase and thymidylate synthetase, inhibiting cell-cycle progression in the S phase.31
Induced Differentiation
HDI-induced growth arrest is able to promote differentiation of leukemia cells, so long as p21 is also present; cells lacking p21 are able to resist the HDI-induced effects of growth arrest and differentiation.32 Furthermore, when patients with APL or the M2 subtype of AML resistant to all-trans-retinoic acid (ATRA) are treated with HDIs, they are able to bypass the inhibition of differentiation caused by their respective PML-RAR-α and AML-ETO fusion oncoproteins.33–35 When HDI and ATRA are used in combination, not only are primary leukemia blasts stimulated to differentiate,36 but APL cells formerly resistant to treatment by ATRA alone become re-sensitized and able to differentiate.35 HDIs can also induce the expression of gelsolin, an actinbinding protein regulating cytostructural changes in differentiation and a putative tumor suppressor.37
Induced Apoptosis
HDIs have been reported to induce apoptosis in several human cancer cell lines, both intrinsically through mitochondrial signals and extrinsically through sensitization of tumor cells to death ligands.38,39 Several HDIs, largely the hyroxamates, can induce mitochondrial permeability transition in leukemia cells, among others,39,40 and in turn initiate a downstream cascade triggering apoptosome activity and the release of caspases 9 and 3.41 Furthermore, treatment with HDIs can attenuate several antiapoptotic proteins in human acute leukemia cells.41 The mechanism underlying apoptosis by HDIs is not yet well understood and seems to vary between cell types. For example, multiple myeloma cells can induce cell death dependent and independent of caspase pathways, but the latter mechanism using calpain activity has yet to be observed in other cell types.42
Histone Deacetylase Inhibitors in Chronic Myeloid Leukemia
HDIs are able to arrest growth and induce differentiation in some cell types while inducing apoptosis in others. While the underlying precisions of the cellular mechanisms and biological effects elicited by HDIs have yet to be characterized, it is understood that targeting epigenetic deregulation in such a manner has great utility in cancer therapy. SAHA is already available on the market as vorinostat (Zolinza, Merck) for the treatment of cutaneous T-cell lymphoma, and there is ongoing research testing the efficacy of HDIs in different leukemic cells.
A phase I trial testing the antitumor activity of the benzamide derivative HDI MS-275 in adults with advanced acute leukemias saw increases in acetylation, p21 expression, and caspase-3 activation, consistent with in vitro data.43 It is suggested that MS-275 (Bayer) be further evaluated for the treatment of less advanced stages of hematological malignancies to better assess drug efficacy. When patients with relapsed or refractory AML or myelodysplastic syndrome (MDS) or newly diagnosed patients were treated with the HDI MGCD0103 (MethylGene), the majority of patients exhibited significant inhibition of HDAC activity within the peripheral blood mononuclear cells, with four of 19 (21%) achieving stable disease.44 With confirmed tolerance of the drug in patients with MDS or advanced leukemias, phase II studies are currently under way.
Recently, hydroxamate analog HDIs were shown to induce acetylation of hsp90, disrupting its chaperone activity and, by association, its interaction with pro-survival proteins (e.g. Akt and c-Raf),45 as well as a number of client proteins known to be oncoproteins in leukemia (e.g. Bcr-Abl). One such hydroxamate is LAQ824, able to deplete messenger RNA (mRNA) levels of Bcr-Abl and decrease protein expression of Bcr-Abl, c-Raf, and AKT, while inducing apoptosis in cells from blastic-phase CML patients and imatinib-resistant cells.46 LBH589 (Novartis) can also induce acetylation of hsp90, and was found to deplete enhancer of zeste homolog (EZH) 2 and DNA-methyltransferase 1 (DNMT1) in leukemic cells, leading to proteasomal degradation of the latter. In addition to modulating acetylation status, LBH589 can induce DNA demethylation, indicative of other areas of epigenetic targeting for antileukemia therapies by HDIs.47
Given that HDIs have the ability to open up the chromatin structure, it makes sense that the increased accessibility to DNA by DNA-targeting agents should be exploited by combination therapies. SAHA is able to downregulate levels of Bcr-Abl and bring about apoptosis in cell lines positive for BCR-ABL and leukemic cells in patients resistant to imatinib; these results were even more enhanced when combined with imatinib, and suggest that this synergistic role may be effective in the imatinib-refractory advanced phases of CML.48 A recent study investigated whether the HDI LAQ824 (Novartis) could induce apoptosis in CD34+ cells from chronicphase CML patients. CML and normal CD34+ cells were cultured with LAQ (10–100nM) alone and in combination with IM (1M) for 96 hours in growthfactor- supplemented medium. Treatment with LAQ824 effectively enhanced histone H3 acetylation in CML and normal CD34+ cells as detected by Western blotting with an anti-histone H3 (Lys9/Lys14) antibody. Moreover, LAQ824 in combination with imatinib was highly effective in inducing apoptosis in CML CD34+ cells. The LAQ824/imatinib combination also resulted in enhanced apoptosis of non-dividing CML CD34+ cells.49
In vitro treatment combining SAHA and dasatinib can increase apoptosis and depletion of Bcr-Abl levels compared with either agent alone, while also attenuating levels of Bcr-Abl escape mutants.50 Similarly, a combination therapy of nilotinib and LBH589 was found to be active against imatinib-resistant CML cells, inducing a greater loss of cell viability in these targets than either agent alone.51 These early data support the in vivo testing of combination therapy in CML patients both sensitive and resistant to imatinib.
The activity of LBH589 has been investigated in a phase Ia/II study in patients with advanced hematological malignancies, including multiple myeloma, MDS, non-Hodgkin’s lymphoma, chronic-phase and blastphase CML, and chronic lymphocytic leukemia. Preliminary data have shown evidence of clinical activity with LBH589 in Hodgkin’s lymphoma, CD4+/56+ hematodermic neoplasm, and AML.52
Summary
With the risk of CML patients developing resistance to the first-line treatment of imatinib, researchers have endeavored to improve the armamentarium against CML. Inhibitors of HDACs have recently been the intense focus of development as compounds against epithelial and hematological cancers. By favoring a more relaxed chromatin structure, HDIs can induce the expression of various genes involved in cellular processes and differentiation, and are quite potent at promoting maturation of leukemic cells. HDIs have also been shown to effectively interfere with co-repressor complexes blocking leukemic cell differentiation such as AML-ETO and PMLRAR- α. Leukemia cells also appear to be susceptible to apoptosis under HDI treatment. How HDIs identify whether to induce differentiation or apoptosis of cancer cells, however, is still not known.
Phase I trials have shown great potential for HDIs in treating hematological malignancies, but currently it appears that the most promise for treating CML lies in the pre-clinical studies in which there is demonstrated synergistic action between HDIs and other targeted therapies in tumor cells. Where imatinib fails due to resistance or lack of response in advanced stages of CML, it is possible that combination treatments can enhance patient outcomes.