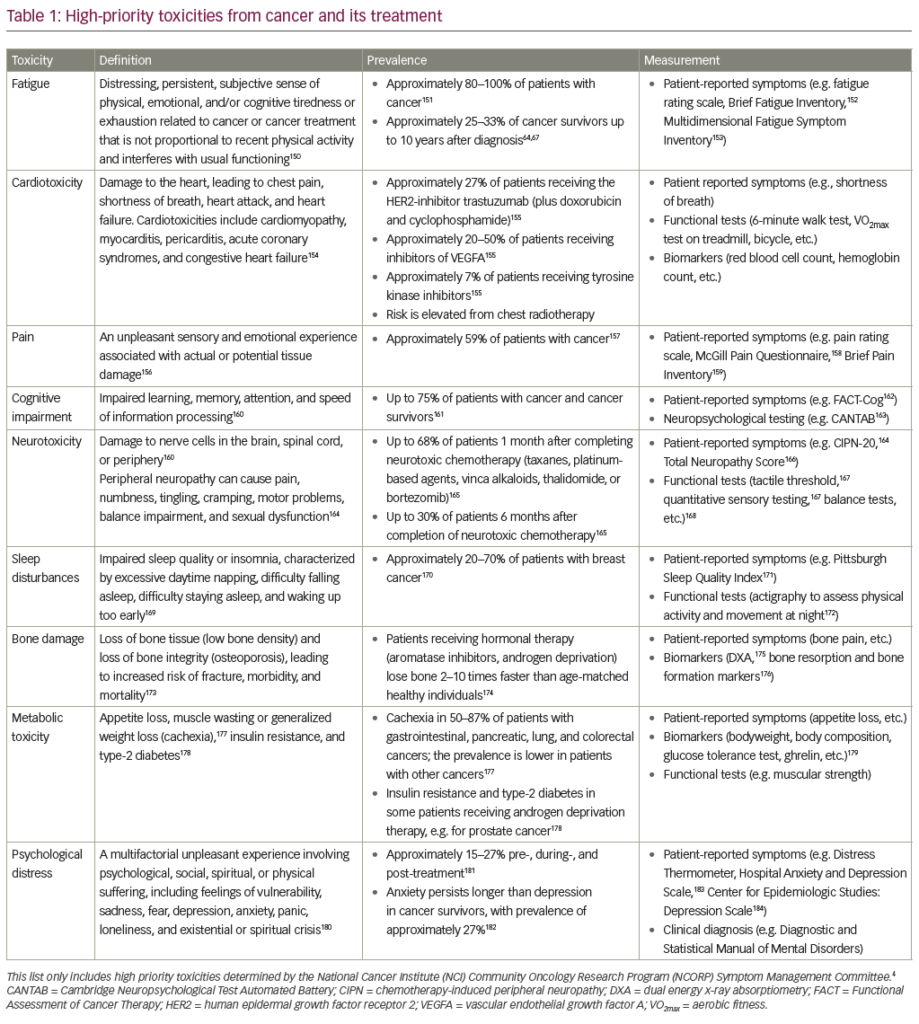
Some patients are primary refractory or develop treatment resistance during the course of the disease, a situation with a particularly poor prognosis and median survival times in the range of only one year.
Over the recent years, there has also been dramatic progress in our understanding of molecular pathogenesis and outcome prediction in CLL. Genetic parameters such as genomic aberrations (11q-, 17p-), the mutation status of the variable segment of immunoglobulin variable heavy chain (VH) genes and surrogate markers (CD38, ZAP-70, LPL) provide prognostic information for individual patients independently of clinical disease characteristics.
Some markers have been associated with rapid disease progression (+12, 11q-, 17p-, unmutated VH, ZAP-70), resistance to treatment (17p-), short remission duration (11q-, 17p-, unmutated VH), and short overall survival (11q-, 17p-, unmutated VH, ZAP-70). These molecular markers are about to enter the stage of risk-stratification for individual patients in clinical trials.
Risk-stratification in CLL
The standard clinical procedures to estimate prognosis in CLL are the staging systems developed by Binet and Rai.1,2 There is heterogeneity in the course of the disease between individual patients within a single stage group. Other markers, such as clinical parameters (age, lymphocyte doubling time), the serum levels of beta 2-microglobuline or thymidine kinase 3,4 and genetic markers of the tumour cells, such as genomic aberrations,5 gene abnormalities (p53, ataxia telangiectasia mutated (ATM) kinase) and the VH mutation status can refine outcome prediction as they may give prognostic information independently of the stage of the disease.6
Genomic aberrations can be identified in about 80% of CLL cases.5 Specific aberrations, such as a deletion 17p- or 11q-, are associated with aggressive disease as evidenced by rapid progression, resistance to treatment (17p-) and short overall survival. Other important molecular genetic parameters to dissect pathogenic and prognostic subgroups of CLL are the mutation status of the VH genes and its surrogate marker ZAP-70. While CLL with unmutated VH and high ZAP-70 expression follows an unfavourable course with rapid progression, CLL with mutated VH and low ZAP- 70 expression often shows slow progression and long survival. Based on VH mutation status and genomic aberrations, patients can be assigned to risk groups with markedly differing survival probabilities6 (see Figure 1). Discordance of ZAP-70 and VH mutation status is observed in 10–25% of cases and may be explained by the presence of additional genetic highrisk features such as 11q-, 17p- or usage of the specific V3-21 gene.7
The molecular prognostic markers such as 11q-, 17p-, unmutated VH and ZAP-70 expression have been identified in retrospective analyses of heterogeneous single-centre patient cohorts and are currently validated in prospective clinical trials. Preliminary data from these trials have led to first concepts of biological risk-stratification in the management of CLL patients.
In routine clinical practice outside trials, the clinical staging systems, the patient’s physical condition and conventional clinical follow-up until disease progression remain the mainstay for the physician’s decision of when and what kind of therapy to apply.
Initial Therapy in CLL
The last 10 years have seen a dramatic development of new agents and combinations for the treatment of CLL. Purine analogues, such as fludarabine (F), were shown to produce superior overall response rates compared to other treatments containing alkylating agents or corticosteroids.8 A trend towards longer overall survival was observed and, subsequently, various combination chemotherapy regimes of different agents, including purine analogues, have been studied.
The most thoroughly studied combination chemotherapy is F plus cyclophosphamide (C). A prospective trial of the German CLL Study Group comparing F versus FC in first-line treatment showed superior response rates for the combination, with a significantly higher remission rate and overall response rate, as well as a longer median duration of response and a longer event-free survival.9 More recently, chemo-immunotherapy has attracted a lot of interest as a treatment option in CLL.
The anti-CD20 antibody rituximab (R) combined with FC chemotherapy (FCR regimen) proved to be highly efficient in first-line treatment. In a recently published study of Keating et al., FCR yielded an overall response rate of 95% and a 69% failure-free survival after four years.10 The FCR antibodychemotherapy regimen is currently being evaluated in a head-to-head comparison against FC in a randomised first-line treatment study. Therapy in Refractory or Relapsed Disease
Unfortunately, all currently available conventional therapeutic options are not curative and eventually all CLL patients will relapse. Patients relapsing after an alkylator-based therapy are likely to respond to an F-containing regimen.
A number of studies have examined the use of the combination of FC in previously treated patients, with the largest series reported from the MD Anderson Cancer Center in Texas, US. Depending on the prior therapy regimen, the overall response rates was 85% in patients who received alkylators as prior therapy and 51% in patients who received a combination of an alkylator with F.11
Once patients become refractory to nucleoside analogue combinations such as FC, outcome is poor, with a median survival time of less than one year. The molecular mechanisms whereby CLL cells become resistant to chemotherapy are poorly understood, but mutations and deletions of the tumour-suppressor gene p53 are more frequent in this situation and are associated with resistance to chemotherapy with alkylators and nucleoside analogues.12
Alemtuzumab, a humanised anti-CD52 monoclonal antibody, appears to have activity against the subgroup of CLL patients refractory to nucleoside analogue combinations and with 17p- or p53 mutation.13 The response rate to the single agent alemtuzumab in refractory patients is in the range of 30–40% with a median overall survival time of approximately 18 months. Clearly, novel treatment options are needed for patients with refractory CLL. One of the most promising agents is the cyclin-dependent kinase inhibitor flavopiridol, which has shown impressive responses in refractory patients. Other promising agents currently being evaluated are oblimersen, an 18-mer phosphorothioate oligonucleotide targeting BCL-2 mRNA, the mTOR inhibitor CCI-779, histone deacetylase inhibitors and the immunomodulatory thalidomide derivate lenalidomide. Clinical trials evaluating novel agents in clinical (refractory) and biological (17p-) high-risk groups of CLL patients are urgently needed.