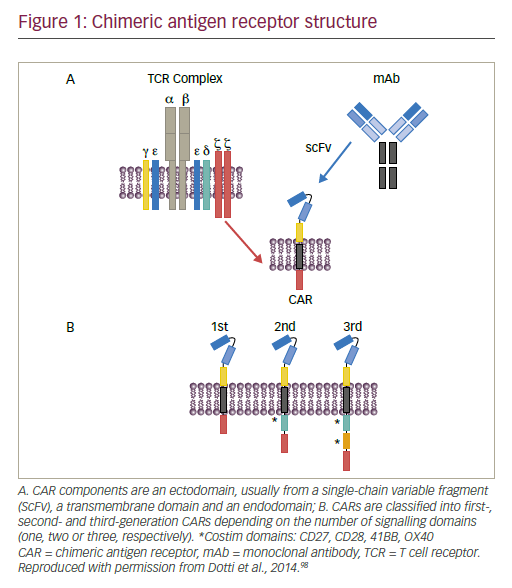
The antitumor activity of immune checkpoint inhibotors in various solid and haematologic malignancies has substantially increased immunotherapy implications for cancer. Once only a niche treatment for selected cancers (renal cancer and melanoma), now cancer immunotherapy has become an important option for many patients, even moving into the first-line systemic treatment setting for melanoma, non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC) and, potentially, a number of other malignancies. Programmed cell death protein 1 (PD-1) inhibitors are already approved in several solid and haematologic malignancies, including melanoma, Merkel cell carcinoma, cutaneous squamous cell carcinoma, hepatocellular carcinoma (HCC), NSCLC, RCC, bladder cancer, Hodgkin lymphoma, head and neck cancer and a broad category of tumours with microsatellite instability high phenotype. Cancers that were once thought to be immune therapy non-responsive, such as NSCLC, are now commonly treated with these options.1
Clinical development and testing of the safety and antitumor activity of anti-PD-1 and programmed cell death ligand 1 (PD-L1) antibodies has been rapid; these antibodies have been shown to be safe across tumours and are effective in a subset of patients in almost every tumour type tested.2 However, while response rates with monotherapy have been high in some tumours, such as melanoma, Hodgkin lymphoma and Merkel cell cancer, single-agent PD-1/PD-L1 inhibition typically achieves relatively low response rates in unselected patient cohorts (an average of 10–30% across trials).2 These response rates can be improved with use of predictive biomarkers, including assessment of PD-L1 expression by tumour and/or immune cells, CD8+ T cell infiltrates in the tumour microenvironment, interferon-gamma (IFN-γ)-centric gene expression profiles and high tumour cell mutational load. Use of predictive markers limits access of these novel therapies, so an alternative is to apply a combination strategy (new checkpoint inhibitor and PD-1 blockade) to increase number of patients benefiting.
In principle, the efficacy of PD-1-blocking therapies might be improved with the following interventions:
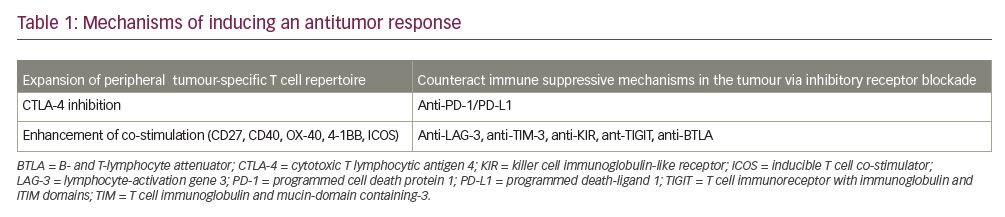
- expansion of the peripheral tumour-specific T cell repertoire (through checkpoint inhibition, engaging co-stimulatory receptors, vaccines, cytolytic viral therapy and cytokines);
- induction of an innate immune response to the tumour (through radiation, cytolytic viral therapy, IFN and toll-like receptor/stimulator of IFN gene pathways); or
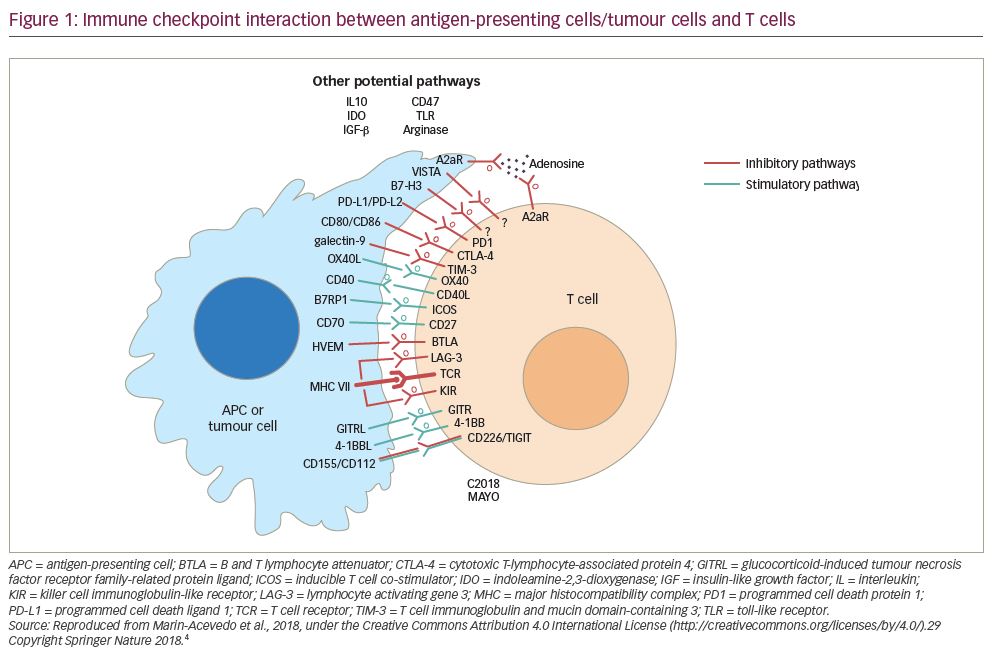
- counteraction of other immunosuppressive mechanisms in the tumour and its microenvironment: depletion of regulatory T cells (Tregs); blockade of inhibitory receptors (T cell immunoglobulin and mucin domain-containing 3 [TIM-3] and lymphocyte activation gene 3 [LAG-3] protein, T cell immunoreceptor with immunoglobulin and ITIM domains [TIGIT], B- and T-lymphocyte attenuator [BTLA]); blockade of macrophage-associated CSF-1R; and depletion of myeloid-derived suppressor cells, adenosine, and vascular endothelial growth factor (VEGF).
The use of single-agent PD-1 pathway inhibition is ideal due to its benefit across various malignancies and tolerable toxicity profile. These facts also make it a desirable backbone for combination approach. Both preclinical and clinical studies have demonstrated additive and/or synergistic effects when distinct immune regulatory pathways are targeted. It is fairly well established that “hot tumours” (tumours with baseline CD8+ T-cell infiltration and IFN-gamma gene signature) are more responsive to PD-1 blockade than “cold tumours”.3 There are number of possiblilties for a tumour to be “cold”, such as: absence of innate immune response to tumour, absence of chemokines to mediate immune response, and/or activation of tumour cell intrinsic pathways leading to immune resistance.4
There are many therapeutic strategies that can increase frequencies of tumour-specific T cells and potentially mediate increased trafficking of T cells into the tumour. They include cancer vaccines, oncolytic viral therapy, co-stimulatory molecule stimulation, targeted therapy, radiation, chemotherapy and adoptive cell therapy (T cells, chimeric antigen receptors). These interventions may, therefore, be particularly useful in tumour types with low or absent response rates with PD-1/PD-L1-directed monotherapy. Combination approaches built on the backbone of PD-1 pathway inhibition that counteract additional inhibitory mechanisms in the tumour microenvironment (such as anti-LAG-2, anti-TIM-3, anti-killer cell immunoglobulin-like receptor [KIR], anti-TIGIT inhibition and anti-BTLA) may be particularly appropriate for T-cell inflamed tumours to enhance or rescue tumour responses achieved with anti-PD-1/PD-L1 monotherapy. Figure 1 is a simplified version of interactions of different immune checkpoints among antigen-presenting cells/tumour cells and T lymphocytes.5
Looking ahead to the future of precision therapy and personalized medicine, selection of appropriate patients for specific combinations may be guided by predictive biomarkers. However, valuable biomarkers for combinations may be distinct from monotherapies, as the experience of tumour PD-L1 expression with PD-1 inhibition versus combined PD-1 and cytotoxic T lymphocytic antigen 4 (CTLA-4) inhibition in advanced melanoma has shown.6
New checkpoint combinations blocking inhibitory receptors
Ipilimumab, an anti-CTLA-4 antibody approved for advance melanoma by The US Food and Drug Administration (FDA), signalled the arrival of modern immunotherapy in solid tumour oncology, further bolstered by later FDA approvals of the anti-PD-1 drugs nivolumab and pembrolizumab. Anti-PD-1 and anti-CTLA-4 agents function to stimulate T cells via different mechanisms, so it was logically hypothesized that combination of these two would result in improved outcomes.6–8 Furthermore, this hypothesis was based on synergy established by preclincal work (B16 melanoma mode and others).7
Based on this evidence, a remarkably successful clinical trial programme combining the α-PD-1 antibody, nivolumab, and the α-CTLA-4 antibody, ipilimumab, demonstrated an initial response rate of >50% in metastatic melanoma; the majority of responses were rapid (evident at the time of the first radiographic assessment at 12 weeks) and durable.6
Anti-PD-1 combined with anti-CTLA-4 has lead to consistent response rates of nearly 60%, superior progression-free survival (PFS) in a number of early- and late-phase trials. Moreover, these data has shown its remarkable stability in subsequent update with 5-year overall survival of 50% that lead to FDA approval of ipilimumab and nivolumab for advanced/unresectable melanoma in 2015.6–8 The impressive anti-tumor activity of ipilimumab and nivolumab has set a fury of clinical trials in a number of other malignancies including NSCLC, RCC, HCC, colorectal cancer, bladder cancer and others. Initial assessments of clinical efficacy appear superior compared with anti-PD-1/PD-L1 monotherapy previously reported in several tumour types. Clinical application of this combination has led to the search for newer partners targeting inhibitory pathways to combine with already well understood molecules (anti-PD-1/PD-L1), some of which are discussed here.
T cell immunoglobulin and mucin domain-containing 3 and lymphocyte activation gene 3 protein
A number of preclinical studies have reported synergy between PD-1 pathway inhibitor and LAG-3 inhibition, as well as TIM-3 inhibition.9–10 Based on this preclinical synergy, a number of early-phase studies are ongoing to demonstrate safety and efficacy of these proposed combinations. A number of early-phase trials are ongoing that evaluate the safety and efficacy of these targets in combination with PD-1/PD-L1 inhibition. Harding et al. conducted a phase I/Ia study in anti-TIM-3 antibody as monotherapy or in combination with anti-PD-L1.11 The results, reported in 23 patients, showed that the combination was well tolerated (most adverse events were less than grade 2; one grade 3 event of anaemia occurred). Although limited efficacy data were reported, clinical activity in relapsed/refractory patients was demonstrated.
LAG-3, another inhibitory immune checkpoint, has been the target of multiple clinical studies in combination with anti-PD-1 agents. For example, Eastgeate et al. reported eftilagimod alpha LAG-3 fusion protein activity in combination with anti-PD-1, pembrolizumab, in advanced/unresectable melanoma.12 In this phase I, dose-escalation study in 18 patients who had previously been treated with pembrolizumab and showed refractory disease, no dose limiting toxicities were observed. Eight out of 16 patients who were eligible for response evaluation had tumour regression.12 However, in a larger phase I/II study of LAG525 (LAG-3 humanized immunoglobulin G [IgG]4 monoclonal antibody [mAb]) +/- spartalizumab (anti-PD mAb), clinical efficacy was modest.13 The combination arm of this study showed dose-limiting toxicity in four patients (grade 3 hyperglycaemia, pneumonitis, brain tumour oedema, fatigue and grade 4 autoimmune hepatitis). There were 12 responses (11 partial responses, one complete response) in patients with solid tumours, whereas therapy was discontinued in 99/121 due to disease progression. Perhaps, relatlimab and nivolumab combination in advanced/unresectable melanoma has the most mature data. Lipson et al. presented data at the American Society of Clinical Oncology (ASCO) annual conference, held in Chicago, IL, USA in May 2021. This study showed median PFS to be significantly longer for combination than for nivolumab alone. Furthermore, this combination was well tolerated.14
Phase II evaluation in selected cohorts is ongoing (ClinicalTrials.gov Identifier: NCT02460224). Ascierto et al. presented initial efficacy data on relatlimab (anti-LAG-3) and nivolumab in anti-PD-1/PD-L1 experienced metastatic melanoma. These data indicate that subjects selected for LAG-3 expression (≥1%) were more likely to respond to this combination. Furthermore, this combination was well tolerated with serious adverse events seen in very few patients (grade 3/4 diarrhoea/colitis in three patients, pneumonitis in two of 262 participants).15 This is a significant improvement over the adverse events associated with the immunotherapy combination (ipilimumab and nivolumab) available in clinic today.
T cell immunoreceptor with immunoglobulin and ITIM domain
T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) is an inhibitory immune checkpoint activated on T cells and natural killer (NK) cells. TIGIT expression is correlated with T-cell exhaustion, decreased circulating T cells, and decreased NK cell cytotoxicity.16 To test the clinical benefit of TIGIT blockade, a number of additional clinical trials targeting TIGIT are in early phases (ClinicalTrials.gov Identifiers: NCT04150965 and NCT02913313). Tiragolumab (anti-TIGIT mAb) was evaluated in a phase Ia/Ib trial as monotherapy, or in combination with atezolizumab (anti-PD-L1 mAb) in advanced solid tumours.17 Seventy-three patients (heavily pre-treated) treated with the combination showed that adverse events were tolerable. However, in phase Ia, there was no objective response; only stable disease was observed in four participants. In phase Ib, there were three observed responses in PD-L1-positive tumours. A subsequent expansion phase is planned in PD-L1-positive tumours.
This early phase study was followed by a randomized, double-blind, phase II study of atezolizumab with placebo or tiragolumab in PD-L1-positive advance/metastatic NSCLC, with primary endpoints of investigator assessed overall response rate (ORR) and PFS. Primary analysis of 135 patients treated on this study was presented at ASCO 2020 and showed an ORR of 16.2% for atezolizumab/placebo versus 31.3% for atezolizumab/tiragolumab, median PFS for atezolizumab/placebo was 3.6 months and 5.4 months for atezolizumab/tiragolumab at median follow-up of 5.9 months.18 With these encouraging results, trials of tiragolumab and other TIGIT-targeting agents are ongoing.
Killer cell immunoglobulin-like receptor
killer cell immunoglobulin-like receptor (KIR) is partially responsible for NK cell activity. Upon binding with human leukocyte antigen-C (HLA-C), KIRs initiate a cascade of activation and inhibitory signals that lead to NK cell inhibition.19 To examine whether the disruption of inhibitory interactions between KIRs and major histocompatibility complex onto NK cells would lead to antineoplastic benefit, Vey et al. studied lirilumab (anti-KIR mAb) in patients with solid and haematological malignancies.19 In this phase I, dose-escalation trial involving 37 patients (solid malignancies n=15, haematological malignancies n=22), no dose-limiting toxicity was observed.19 The most common adverse events noted were pruritus 19%, asthenia 16%, fatigue 14%, infusion-related reaction 14% and headache 11%; however, most were mild to moderate. In 22 (with evaluable disease at baseline) of 37 patients, no objective response was observed. In all 37 patients included, 15 showed progressive disease and 22 had stable disease. There appeared to be a PFS benefit in breast cancer, ovarian cancer and acute myeloid leukemia. These informative data have paved the way to combination strategies with partners like anti-CTLA-4 and/or anti-PD-1/PD-L1.
B and T lymphocyte attenuator
B and T lymphocyte attenuator (BTLA/CD272) belongs to the CD28 family and is structurally related to CTLA-4 and PD-1. Interaction of BTLA and herpes virus entry mediator has been demonstrated to be inhibitory on T cells.20 Furthermore, increased expression of BTLA conveys poor clinical outcome, and blockade of BTLA (via various mechanisms) translates into clinical benefit. These data are essentially preclinical at this point, and clinical trials are in progress to establish hypothesized benefit.20
Newer checkpoint using co-stimulatory receptor agonists
Co-stimulatory receptors, including CD137 (also known as 4-1BB), glucocorticoid-induced tumour necrosis factor receptor (GITR; CD357), CD40, OX40 and CD27, are predominantly expressed by activated T cells, but can also be upregulated by activated NK cells, Tregs and other immune cells.21 Agonistic antibodies may promote NK cell-mediated antibody-dependent cellular cytotoxicity.22 Stimulation of co-stimulatory receptors can increase T cell frequencies in the periphery, modulate T effector functions, reverse T cell anergy in the tumour, counteract the suppressive activity of Tregs, and enhance cytokine secretion by NK cells.
4-1BB
Synergy between PD-1 and CD137/4-1BB activation has been demonstrated preclinically.22,23 Urelumab, an agonistic CD137 antibody, has been in clinical development for several years. Initial clinical trials were stopped due to hepatotoxicity, leading to substantial dose reduction in later trials. Furthermore, aggregated analysis of three studies showed that a dose of 0.1 mg/kg every 3 weeks carried the immune stimulation function and had a safety profile that was acceptable.24 In particular, doses greater than 1 mg/kg showed significant incidence of hepatotoxicity.
The combination of urelumab with the α-PD-1 antibody nivolumab has been studied in NSCLC, squamous cell head and neck cancer and lymphoma.25 Initially, reported response rates in these four cancers were modest and not higher compared with historical controls of PD-1 monotherapy. A second CD137 agonist, utomilumab, has also been evaluated in phase I study as a single agent.26 In this study, 55 patients were evaluated for safety, tolerability, pharmacokinetics, preliminary clinical activity and pharmacodynamics. The ORR was found to be only 3.8%; the incidence of hepatotoxicity was much lower at the doses tested. From these two studies, it seems that CD137 is a suboptimal target for a single-agent approach; perhaps a combination with anti-PD-1/PD-L1 will yield the desired responses in the first-line setting or in the refractory setting.
Glucocorticoid-induced tumour necrosis factor receptor
GITR is a co-stimulatory molecule expressed primarily by Tregs, effector T cells and NK cells, and inhibits the suppressive activity of Tregs. AMG 228, an agonistic human IgG1 mAb that binds to GITR, was studied in the phase I setting in heavily pre-treated solid malignancies.27 This 30-patient study included metastatic colorectal cancer, head and neck squamous cell carcinoma, RCC, NSCLC and melanoma. Overall, AMG 228 was well tolerated, with most adverse events being low grade. However, 12 patients (40%) had serious adverse events that were considered treatment related, and there was one death due to pneumonitis related to AMG 228.27 Additionally, there was modest antitumor activity with single-agent therapy. Combination strategies with anti-PD-1/PD-L1 are currently being evaluated in multiple clinical trials.
OX-40
Another co-stimulatory molecule of interest, OX-40, which is primarily expressed on activated T cells, has been in clinical development as a single agent or in combination with anti-PD-1/PD-L1 backbone. Hamid et al. presented data on a first-in-human study of PF-8600 as a single agent, an OX-40 agonist, fully-human IgG2 mAB. The reported data, in a small number of patients, demonstrated tolerability and some clinical activity. The most common adverse event reported was fatigue (33%). Four of nine patients had the best ORR of stable disease.28 Follow-up combination studies with anti-PD-1/PD-L1 are ongoing.
CD40
CD40 is a co-stimulatory molecule, present in a variety of immune and non-immune cells, is thought to be essential to initiate specific immune response by activating antigen-specific naïve B and T cells.29 A number of solid malignancies (bladder, melanoma, breast, lung, colon and B-cell malignancies) are known to express CD40 to differing extents, and different antibodies directed at CD40 are in clinical trials with limited data available. Most data focus on safety and tolerability. A combination approach of anti-CD40 with commercially available checkpoint inhibitors has yielded some interesting results with slightly improved response rates over single-agent checkpoint inhibitors.30 Other trials testing CD40 agonism in combination with either chemotherapy or immunotherapies are in progress, and data are awaited eagerly.
Inducible T cell co-stimulator
Inducible T cell co-stimulator (ICOS/CD278), expressed by T cells, plays important role in memory and effector T cell development and response. A number of anti-ICOS agonists are in various phases of clinical trial. Currently available data show that this approach is relatively safe, with adverse events (mostly grade 1/2) including fatigue, fever, transaminitis and diarrhoea. Efficacy data of the single agent or in combination with anti-PD-1/PD-L1 or CTLA-4 is not yet available,31 but a number of trials are ongoing.
CD27
CD27, another co-stimulatory molecule, has entered the clinical picture with early data showing clinical activity as a single agent and in combination with anti-PD-1/PD-L1.32 However, caution is advised, since most of the trials are in their early phase and outcome data reports are not available.
Conclusion
As long-term data on approved immune checkpoint inhibitors have matured, so has their clinical application in various malignancies. There still remain certain malignancies that fail to respond to current checkpoint inhibitors or become refractory, and newer checkpoint inhibitors are the hope for these patients. The data discussed show that, as single agents, these new checkpoint inhibitors have little benefit; their most appropriate partners appear to be PD-1/PD-L1-targeting agents. A vast number of trials are in progress and are trying to harness the benefit of these new combinations. It would be wise to be cautious, since most of the data available are in their infancy. Additionally, hope is that data on these newer checkpoint inhibitors will bring along predictive biomarkers to identify patients who will most benefit, or help in deselecting patients who will have significant toxicities. A more auspicious hope is that combinations of newer checkpoint inhibitors with PD-1/PD-L1 inhibitors or other partners will have a more tolerable profile as well. In addition to these newer checkpoint inhibitors, data in solid malignancies also looks promising, such as oncolytic viruses, with combinations of toll-like receptor agonists or histone deacetylase inhibitors with either anti-PD-1/PD-L1 or anti-CTLA-4 agents.