Therapeutic cancer vaccination is a promising strategy that aims to treat late stage disease by using the patient’s own immune system.1 The immune system has the potential to eliminate cancer cells, since cancer cells express antigens that are unique to the tumour or are over-expressed in the tumour. However, despite many years of research, the effective induction of strong and durable antitumour T-cell responses in vaccinated patients remains a challenge.1 The efficient presentation of cancer antigens to T cells is key to successful vaccination; mouse models have indicated that the generation of protective antitumour immunity depends on the presentation of tumour antigens by dendritic cells (DCs).2
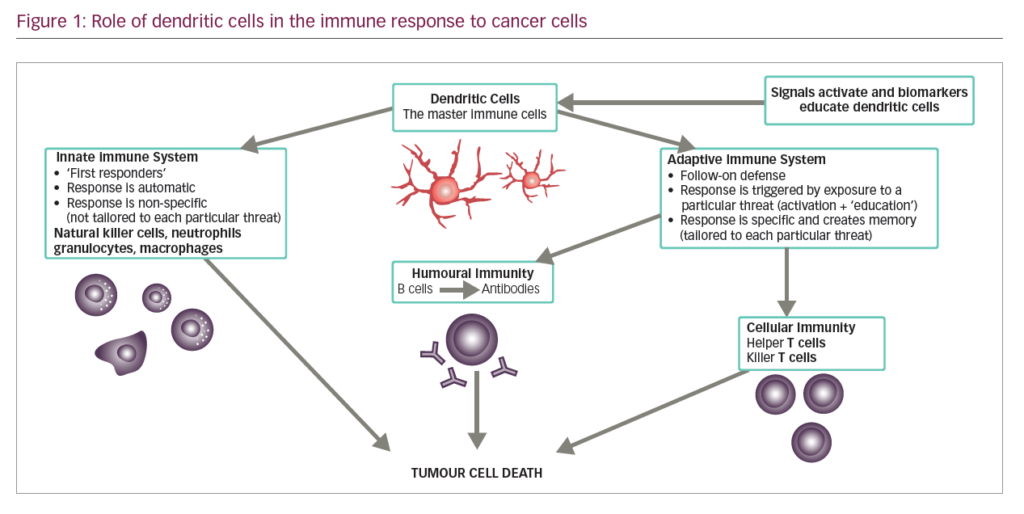
DCs are leukocytes derived from bone marrow and are an important component of the adaptive and innate immune system (Figure 1). They induce an immune response by presenting captured antigens to naïve T cells in lymphoid tissues.3 In addition to their immune stimulatory functions, DCs infiltrate most human tumours, where they have regulatory effects including both tumour growth inhibition and stimulation of the immunosuppressive tumour microenvironment.2 As a result, DCs have been investigated as potential therapeutic tools in cancer.4–12
DCs can be derived from monocytes or CD34+ stem cells, for example by direct isolation of peripheral blood mononuclear cells (MNCs).13,14 Autologous DCs can be generated ex vivo, activated, loaded with tumour antigens and then returned via injection to the patient with the aim of eliciting tumour-specific effector T cells that kill tumour cells and induce immunologic memory to control tumour relapse (DC vaccination).15,16 The first DC vaccination study was published in 1996 and used DCs generated ex vivo from peripheral blood monocytes to immunise B-cell lymphoma patients.17 Since then, numerous small clinical trials have evaluated DCs as cellular mediators for therapeutic vaccination of patients with cancer, many demonstrating a survival benefit.4–12 These studies have included a number of different clinical indications including: prostate cancer,18 colorectal cancer,19 melanoma,12,20–23 ovarian cancer,7,24 glioblastoma,8,11,25–27 renal cell carcinoma,9 pancreatic cancer28 and haematological malignancies.10 In 2010, the US Food and Drug Administration (FDA) licensed Sipuleucel-T for clinical use. This was the first DC-based vaccine to be approved having shown prolonged overall survival among men with metastatic castration-resistant prostate cancer29,30 and this technology has attracted considerable interest as a therapeutic option in multiple cancer types. However, while many vaccines have elicited tumour-specific immunity, objective clinical response rates remain low.
Improved techniques for harvesting DCs have resulted in many new and novel vaccine strategies being developed. This review article aims to explore the characteristics of DCs that make them an ideal choice for anti-tumour vaccines, as well as providing a detailed description of the processes and discussing the technical challenges of DC vaccine production.
Characteristics of dendritic cells
DCs are uniquely suited to cancer immunotherapy as they form a network of antigen presenting cells (APCs) that induce primary immune responses, improve the effector functions of previously primed T-lymphocytes and facilitate cross-communication between innate and adaptive immunity (Figure 1).31 DCs are extremely efficient APCs and responses can be achieved from low numbers of T cells since their potency for inducing T cell proliferation is 10–100 times that of B-cells or monocytes.31,32 They sensitise antigen-specific responses in both CD4+ and CD8+ T cells. The latter differentiate into cytotoxic T-lymphocytes (CTLs), conferring direct anti-tumour effects.31
The other feature of DCs that makes them ideally suited for clinical use is the fact that they can be differentiated in vitro from CD34+ progenitors from purified cord blood, bone marrow or cytokine-mobilised peripheral blood progenitor cells,33 or isolated in low numbers in peripheral blood, which can be enhanced following pre-treatment with a DC-stimulating factor such as Flt3 ligand.16,34 The largest numbers of DCs can be obtained with the use of specific cytokines to mobilise CD34+ cells from the bone marrow to the bloodstream.35 Substantial savings in materials and other costs can be achieved if monocyte-derived DC for multiple treatments are generated from cryopreserved monocytes rather than from fresh monocytes.36
Generation of dendritic cell-based vaccines
As a result of the immunosuppressive nature of most tumours, T cells activated by vaccination may become inactivated at the tumour site. High DC numbers and multiple doses of vaccine are therefore needed to assure that sufficient activated CTLs are generated to eliminate tumour cells and ensure that sufficient T cells enter the memory compartment to act as rapid responders in case of tumour relapse. A typical DC vaccination dosage is 5–50 million DC cells, and up to 15 vaccinations may be needed.12,37 The number of cells needed for immunotherapy necessitates their standardised production on a scale large enough to sustain repeated vaccinations while conforming to current good manufacturing practice (GMP) guidelines. Therefore, the generation of DC vaccines involves a number of key stages (Figure 2). Here, we elaborate on the different stages that are key for a successful DC vaccination.
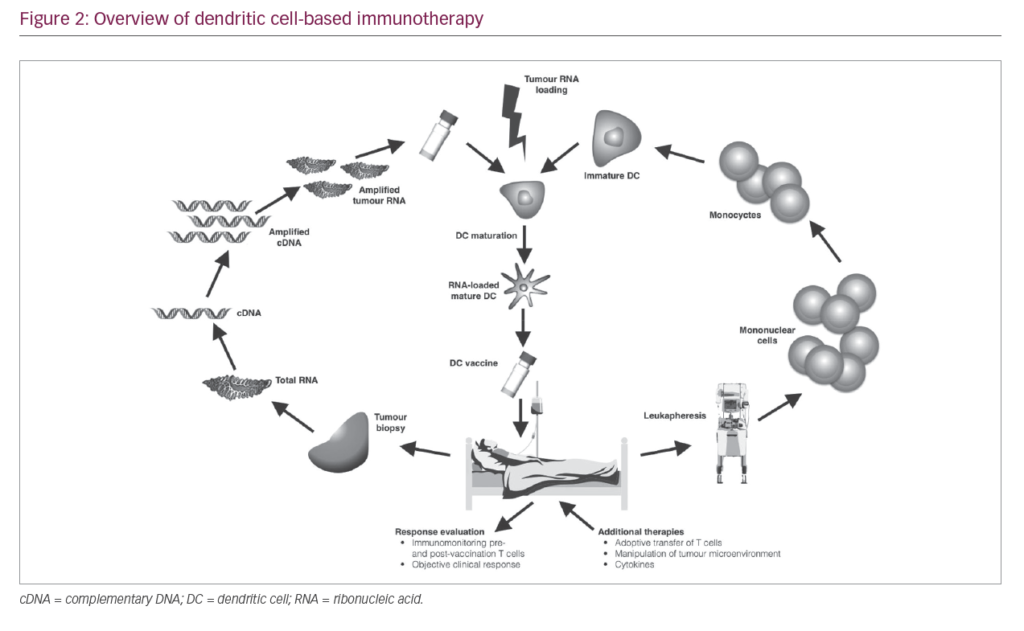
Harvesting
Numerous techniques have been used to isolate DC precursors; these include apheresis, elutriation, filtration, adherence, positive selection of CD14+ monocytes and negative selection via lymphocyte depletion.13 However, harvesting by leukapheresis can generate a large number of DCs and has become the standard procedure for collection in many DC-based clinical studies.13
A number of apheresis systems are available for the harvesting of DCs. In 2003, a study compared MNC programmes on two apheresis systems (COBE® Spectra [Terumo BCT, Lakewood, Colorado, USA] and AS.TEC [Fresenius Kabi, Bad Homburg, Germany]).38 The study found that the AS.TEC cell separator showed significantly better monocyte collection rates and efficiency compared with the COBE Spectra system in 5 L collections (medians of 11.0% versus 7.4% [rate] and 51.9% versus 31.9% [efficiency], respectively; all p<0.05), but not in 10 L collections (10.4% versus 8.5% [rate] and 67.6% versus 55.2% [efficiency], respectively; all p≥0.21).38 In both 5 and 10 L collections AS.TEC resulted in significantly higher residual red blood cell (RBC) yields compared with COBE Spectra (5 L: 227 versus 84 x 109/bag, respectively; 10 L: 478 versus 142 x 109/bag; all p<0.001).38 To optimise the procedures, different mononuclear programme settings for COBE Spectra and AS.TEC were subsequently evaluated. For all programme settings the mean MNC purity was ≥93%.39
However, use of the standard MNC programme of both devices resulted in significantly higher collection efficiencies of CD14+ monocytes, CD3+ cells, CD4+ cells, CD8+ T cells, CD16+ CD56+ natural killer (NK) cells, and residual platelets (all p<0.01) compared to the modified programme settings, due to a higher centrifuge speed.39 A 2005 study also compared two different settings of the COBE Spectra apheresis system: the MNC and AutoPBSC settings.40 Both settings produced similar mean yields of leucocytes (4.24 and 4.52 x 109/unit, respectively) and lymphocytes (T cells: 2.05 and 2.16 x 109/unit).40 However, products derived using the AutoPBSC setting contained significantly (p<0.05) greater mean yields of granulocytes (0.11 versus 0.20 x 109/unit), monocytes (0.98 versus 1.13 x 109/unit) and RBCs (50.9 versus 63.5 x 109/unit), but significantly fewer platelets (mean yield: 215 versus 118 x 109/unit).40
Different programme settings have also been evaluated for the COM.TEC® cell separator (Fresenius Kabi, Runcorn, UK).41 In 2006, twenty-four 5 L MNC collections were performed from non-cytokine-stimulated donors. The MNC setting resulted in a high mean CD14+ collection efficiency (84%) and mean cell yield (1.2 x 109/unit), but high residual platelets (mean efficiency: 42.7%; mean yield: 317 x 109/unit). The use of a dual-stage chamber in the PBSC-LYM setting produced fewer residual platelets (mean efficiency: 9.8%; mean yield: 79 x 109/unit), but did not achieve the target mean CD14+ yield of 1 x 109 cells.41
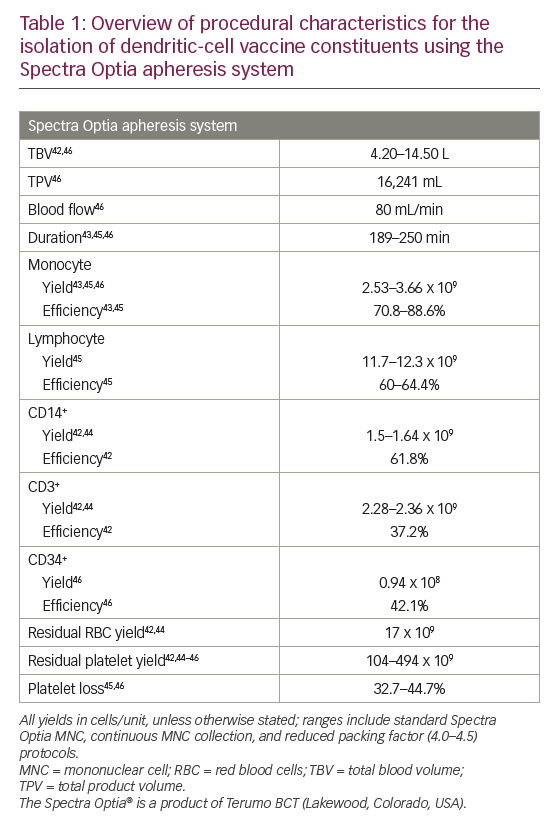
More recently, the Spectra Optia® apheresis system was developed (Terumo BCT). In a 2013 study, 12 MNC collections were performed using this system in non-cytokine-stimulated donors.42 This resulted in satisfactory MNC yields (including a median CD14+ of 1.28 x 109/unit) with low residual RBC and platelet yields (mean: 17.0 and 104 x 109/unit, respectively).42 A 2014 study also showed that this system was able to isolate and enrich a highly purified MNC fraction (88.6% monocytes).43 In a head-to-head study comparing Spectra Optia with other apheresis systems (COM.TEC and Amicus™ [Fresenius Kabi]), all devices resulted in similar total MNC yields.44 The Spectra Optia device, however, collected significantly more CD14+ monocytes than the Amicus device (mean yield: 1.64 versus 1.20 x 109; p=0.002) and significantly fewer CD3+ cells (not required for DC generation) than the COM.TEC device (mean yield: 2.36 versus 3.03 x 109; p=0.002). The Spectra Optia products had the lowest mean RBC content (yield: 16.8 versus 53.0 [Amicus] and 124 [COM.TEC] x 109; all p≤0.002) while the Amicus products had the lowest mean platelet content (yield: 46.2 versus 114 [Spectra Optia] and 614 [COM.TEC] x 109; all p<0.001).44
Further development of the Spectra Optia system to perform continuous MNC collection, enabling shorter procedure times with reduced product volume, showed similar (all p≥0.05) mean yields of monocytes (2.91 versus 3.52 x 109) and lymphocytes (11.7 versus 12.3 x 109), as well as contaminating platelets, RBCs and granulocytes compared with the standard Spectra Optia MNC system.45
Regarding the collection of other cells, a recent retrospective study compared the Spectra Optia MNC and COBE Spectra MNC systems for autologous PBSC collection and found no significant differences in the median CD34+ cell content of collected products (0.61 × 108 versus 0.94 × 108), CD34+ cell collection efficiency (43.5% versus 42.1%), and loss of platelets (40.1% versus 44.7%).46
Overall, modern apheresis systems have shown to be broadly similar in terms of performance and efficacy (MNC/monocyte yields and collection efficiencies).38,44 However, the Spectra Optia system has shown some benefits over other systems with regard to the collection of CD14+ cells required for DC generation, and minimising RBC and platelet contaminants.44 Platelet contamination is a problem when monocytes are purified by adherence due to space competition as they bind to the monocytes,47 forming clumps and reducing monocyte yield.38 To address this, reduction of the preset buffy coat volume has been shown to optimise leukapheresis harvests using the COBE Spectra system and AutoMNC setting.48 Similarly, reducing the packing factor (from 4.5 to 4.0) used in the Spectra Optia continuous MNC device has been shown to further reduce platelet contamination (mean total product 315 versus 494 x 109, respectively; p<0.001) while retaining the mean monocyte yield (2.67 versus 2.69 x 109; p≥0.05).45
Enrichment
A number of different approaches have been used for monocyte enrichment. Methods of enrichment include plastic adherence, positive selection using magnetic beads coupled to CD14 antibodies or cell depletion using beads coupled to antibodies against CD2+ T cells and CD19+ B cells to remove non-monocytes. Available systems for these procedures include the CliniMACS® cell-selection system (positive selection; Miltenyi, Bergisch Gladbach, Germany) and Isolex 300i Magnetic cell selector (immunomagnetic cell depletion; Nexell, Irvine, California, USA). In general, positive selection can provide a high purity of immature DCs with a low yield (e.g. 97 ± 1% and 8 ± 3%, respectively; CliniMACS),49 whereas removal of non-monocytes provides a low purity, but a high yield (42 ± 10% and 21 ± 6%, respectively; Isolex 300i).49 Conventional plastic adherence procedures tend to provide more balanced purity and yield (72 ± 4% and 25 ± 5%).49
A study by Wong et al. also showed that it is possible to enrich monocytes by counterflow centrifugal elutriation (Beckman J-6M centrifuge; Beckman Instruments, Palo Alto, California, USA) with a similar DC purity and yield obtained using the Isolex 300i system (67–80% and 11–13%, respectively).50 The Elutra™ system (Terumo BCT) is a closed (thereby minimising the risk of contamination) counterflow centrifugal elutriation cell separation system that can also isolate monocytes from other blood components for DC generation. Monocytes are collected from the elutriation chamber in the last fraction, which also contains granulocytes, and in general the purity of the monocytes isolated using the Elutra system is at least 80%.51,52 The presence of up to 16% granulocytes in the monocyte product have been found to have no significant effects on the quality of the DCs formed. However, once the concentration of granulocytes increases above 16%, a gradually increasing detrimental effect on DC quality is observed in terms of the migratory capacity of DCs and lower expression levels of CD80, CD40 and CD86.51 Nevertheless, the presence of 20–30% granulocytes in a monocyte product has been shown to have no major influence on the quality of the DCs generated from monocytes.51 In conclusion, as the Elutra system isolates monocytes with a purity of ≥80% and can isolate up to 7 × 109 MNCs,52 it is a suitable system for the large-scale isolation of monocytes for the generation of DCs.
Differentiation and maturation
Differentiation of monocytes into immature DCs typically involves culture with granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4 or -13, which lead to the formation of immature DCs.53 Maturation of cells is then required since mature DCs migrate more effectively than immature cells and induce better anti-tumour responses.54,55 A maturation cytokine cocktail containing IL-1beta, tumour necrosis factor (TNF)-alpha, IL-6 and prostaglandin E2 (PGE2) has been shown to yield mature DCs with high migratory potential.56 However, there is some evidence that IL-6 and PGE2 may induce tolerogenic DCs and may even promote programmed cell death protein ligand (PD-L)1 expression, which plays a major role in tumour escape from the immune system.57,58 Indeed, some studies have shown that differentiation of bone marrow-derived DCs with GM-CSF and IL-15/IL-14 or FMS-like tyrosine kinase 3 ligands (FLT3L), as well as suppression of the PGE2 pathway, can produce more mature and immunogenic DC phenotypes than standard DC preparations.59–62 While differentiation with GM-CSF/IL-4 has been shown to preferentially expand the conventional DC type 2 subset and produce more allogeneic DCs and inflammatory mediators than FLT3L, the latter expands both conventional DC type 1 and 2 subsets and results in more efficient migration of DCs to lymph nodes with a separate cytokine profile.60–62 Overall, GM-CSF/IL-4 DC resemble inflammation-emergent DCs whereas FT3L DCs represent steady-state resident DCs.61 Other cytokine cocktails including toll-like receptor ligands to induce immunostimulatory cytokines are also under evaluation.63
Various protocols for differentiation and maturation have been developed over the years. Early generation of mature DCs from human monocytes in vitro required differentiation with GM-CSF and IL-4 for 5–7 days, followed by 2–3 days of activation. In 2003, a protocol was developed for differentiation and maturation of monocyte-derived DCs within 48 hours using in vitro culture, which was termed FastDC. Not only does a shorter protocol save labour, cost and time, but it also more closely models the differentiation of DCs in vivo.64 Monocytes were incubated for 24 hours with GM-CSF and IL-4, followed by activation with proinflammatory mediators for a further 24 h.64,65 This protocol was later adapted for large-scale clinical use, which involved collection of MNCs by apheresis, isolation of monocytes by the Elutra Cell Separation System, and culturing in sterile Teflon bags.66 This manufacturing method has now been transferred into a closed-system (to minimise the risk of contamination), validated, GMP-compatible protocol, which may pave the way for larger multi-centre clinical trials.67 Overall, with its shorter incubation times, lower risk of contamination and large-scale production, FastDC is perhaps the most appropriate protocol in most situations; as a result it is widely used in clinical practice.
Antigen loading of dendritic cells
Following collection and enrichment, DCs must be loaded with antigens. Tumour antigens are frequently inconsistent in their expression between and within tumours, therefore loading DCs with peptides derived from multiple antigenic proteins improves the chance of recognition.53,68 Human DCs may be loaded with whole tumour-associated antigens; the most widely used method is incubation with human leukocyte antigen (HLA) class I- and class II-binding peptide. However, one potential problem with protein loading is the limited half-life of antigen expression on the DC surface.53 An increasingly used alternative approach is to transfect DCs with tumour RNA-encoding antigens that, via continuous gene expression, can maintain antigen expression for the life of the DC.53 Examples include AGS-003, which has high transfection efficiencies and is well tolerated with only mild injection site reactions.9 When specific tumour antigens are unknown, other DC loading strategies may be used; these include pulsing with the beta-galactosidase protein, H-2K restricted peptide, tumour cell lysates (prepared using freeze-thaw cycles; promising results have also been observed with squaric acid treatments of cell lysates69), and irradiated tumour cells and electrofusion of DCs with tumour cells.70 Of these, there is evidence that DC–tumour fusion cells may provide the most effective vaccines to treat existing tumours.9
More recently, it has been shown in a preclinical study have shown that novel antigen-capturing nanoparticles can deliver tumour-specific proteins to APCs, improving the efficacy of anti-PD-1 therapy in murine models of melanoma.71 This potentially offers a new strategy for the synergistic improvement of cancer immunotherapies that could be applied to the antigen loading of DC vaccines.71 In situ vaccines are also in development, which target DCs within the tumour microenvironment through intratumoural injections of toll-like receptor ligands or FLT3L, resulting in recruitment and local activation of DCs in the tumour.72 Overall, the choice of procedure depends on both the knowledge of antigens expressed by the tumour, and the availability of tumour tissues or other clinical grade sources of antigen.53
Administration of dendritic cell-based vaccines
Administration of DC vaccines is simple and the clinical resources required are minimal; a typical procedure involves thawing the cells for administration and subsequent injection of the cells. However, the success of DC-based vaccines depends on accurate delivery of cells. Delivery is typically intravenous,73,74 but subcutaneous,73 intradermal,74,75 intranasal,74 intraperitoneal,74,76 intratumoral77 and intranodal75 injection modes have been evaluated.73–75 Effective migration of DCs to the lymph nodes is essential in advanced cancer to interact with naïve T cells, but DCs tend not to migrate easily. Intravenous administration may be quick and simple; however it may allow preferential retention of some cellular therapeutic agents in the liver.78
Similarly, subcutaneous or intradermal routes may allow only a modest fraction of DCs to enter the lymph nodes, and the long migratory times may reduce the ability of DCs to produce the required cytokines and chemokines.78 To overcome this, intranodal delivery has been attempted in melanoma patients with mixed success, but this route can be technically challenging and may lead to DC accumulation in perinodal fat tissues and lymph node damage.75,79,80 Recent data suggest that the prolonged (up to 7.5 weeks) delivery of DCs through implantable lymphatic delivery ports is feasible and tolerable, though the potential advantages of this route of administration are yet to be confirmed.78
It is clear that further research is required to optimise the route of administration for DCs in specific indications and/or aid migration of DCs to the lymph nodes. Indeed, various methods are currently available to stimulate DC migration to the lymph nodes, including conditioning tissues using inflammatory cytokines,81 prostaglandins,82 chemokines,83 and matrix metalloproteinases.84 In vivo migration of magnetically labelled DCs can be tracked using magnetic resonance imaging to locate the cells in vivo. When used in vivo, this method can detect very low numbers of DCs, is safe and its use is feasible in humans.80 As the routine imaging protocols involved are readily available on conventional magnetic resonance imaging systems, it is hoped that this process may enable investigators and clinicians to obtain a deeper understanding of cellular treatment modalities and determine their respective efficacy.80
Quality control measures for dendritic cells
The clinical utility of DCs is dependent on effective means of quality control assessment. Therefore, there is a need for rapid methods to assess the product consistency related to the manufacturing process, as well as the identification of reliable quality assurance markers. The expression of classical DC markers, such as CD80, CD86, CD83 and positivity to CCR7,85 can be measured using flow cytometry, but this is of limited use since these markers are expressed by DCs with different phenotypes. Gene expression profiling can be used to test the degrees of product consistency related to the manufacturing process and variability due to intra- and inter-donor factors, and how each factor affects single gene variation. For each gene, an index of variation is calculated and used to select candidate markers for identity testing, and also to define a set of genes that may be useful comparability and potency markers. This technique has proven effective, and has identified 29 potential markers for characterising DCs.85
Potency testing is useful to determine the functional capacity of the DC product, and the FDA recommends the development of potency assays for characterisation of cellular products used for human therapy. An IL-12p70 production assay has been developed that is applicable to small or large samples of DC vaccines generated under different conditions. The assay measures the ability to secrete IL-12p70 and respond to helper T cell signals (CD40L). It then quantifies IL-12p70 using an immunobead multiplex platform. The assay is reproducible, robust, and cost-effective, and can discriminate between DCs matured in the presence of different cytokine cocktails and between DCs obtained from healthy donors and patients with human immunodeficiency virus-1 or cancer. It is being used in ongoing early-phase clinical trials to determine its utility in predicting the in vivo efficacy of DC-based vaccines.86 Another novel method, the COSTIM bioassay, selectively measures co-stimulatory activity, or functional potency of the DCs. T cells are stimulated with a sub-optimal amount of anti-CD3 antibody and are unable to proliferate unless a source of co-stimulation (DCs) is added to the culture.87 In addition, the capacity to induce alloresponse (mixed lymphocyte reaction) is currently used to determine the potency of DC vaccine.
Clinical application of dendritic cell-based vaccines
Numerous DC-based anticancer therapeutics are in clinical development.88 Purified populations of CD1c (BDCA-1+) myeloid DCs were manufactured by pulsing autologous CD1c+ DCs, prepared by magnetic bead immunoselection from apheresed peripheral blood MNCs, with a cocktail of HLA-A*0201-restricted peptides and keyhole limpet haemocyanin, a potent immune-stimulating glycoprotein. The resulting vaccine was well tolerated in a phase I feasibility study in 12 patients with advanced metastatic prostate cancer, with preliminary data showing a mean survival of 18 months for 11/12 patients and one patient remaining alive after over 5 years.89 In addition, some evidence of the induction of immune responses to control antigens was also observed (anti-keyhole limpet hemocyanin antibodies in 25% of patients, delayed-type hypersensitivity to influenza matrix protein in 25% of patients).89
In patients with melanoma, Ridolfi et al. have reported a beneficial effect on survival using DCs prepared from peripheral blood MNCs using leukapheresis, adherence purification and pulsing with autologous tumour lysate; the median overall survival was 16 months, increasing to 22.9 months in patients who showed positive results in delayed-type hypersensitivity testing.21 Thus, there is a need for treatments aimed at improving immunological responsiveness to the vaccine, such as pre-leukapheresis radiotherapy and/or the addition of interferon alpha to the vaccine with the aim of increasing the vaccine-induced tumour immune response in patients with metastatic melanoma.90
More recently, naturally circulating DCs were used to vaccinate advanced melanoma patients.91,92 Briefly, this method involves a highly standardised rapid isolation procedure with antibody-coated magnetic beads that produces clinically applicable purified DCs without an extensive culture period, which may have a positive effect on the immunological capacity.91,92 Once isolated from the patient and cultured overnight, the purified DCs are then activated, loaded with tumour-associated peptides and returned to the patient via intranodal injection. Results of two small studies (n=15 and n=14) in patients with advanced melanoma have shown this approach to be feasible, with the induction of favourable immune responses (33–47% showing tumour-antigen-specific CD8+ T- cells in blood), minimal toxicity, evidence of improved progression-free survival versus controls (4.0 versus 2.1 months) and even objective tumour responses in two patients.91,92 As with the previous melanoma studies, future phase III trials are needed to investigate the potential survival benefit.
Future perspectives
DC vaccines present a variety of opportunities for combination therapy with other complementary immunological cancer treatments to enhance the initial anti-tumour effect, as well as protecting against future remissions. This is because of their central co-ordinating role in both the innate and adaptive immune systems; in particular, their use in combination with checkpoint inhibitors could synergise both immune responses and the inhibitory pathways developed by cancer cells to evade immune responses.2 Other combined treatment approaches with DC vaccines include modulation of angiogenic pathways, depletion of soluble inhibitors, and adoptive transfer of T cells, which may be genetically modified to resist T cell-directed tumour-immune evasion strategies.53
The use of DCs in cancer therapy offers the potential of personalised medicine; a DC vaccine loaded with tumour-specific amino acid substitutions (neoantigens) is being investigated in patients with advanced melanoma.93 However, a major disadvantage of this form of immunotherapy is the requirement for vaccines that are tailor-made for each individual (patient-specific). This is a substantial technical challenge involving large scale production and extensive resource utilisation to prepare every individual course of treatment.
Universal implementation of the technique will only be possible if more cost-effective and efficient strategies are developed. These challenges may be resolved with novel methods that are under investigation, which will target antigens to distinct DC subsets and simultaneously employ adjuvants to activate these cells to induce immunity.94,95 These subsets include monocyte-derived DCs, conventional DC type 1, conventional DC type 2, and plasmacytoid DCs.96 Monocyte-derived DCs are most frequently used for vaccination purposes, based on technical aspects such as their availability and in vitro expansion.96 However, recent technological developments have enabled the efficient isolation of conventional and plasmacytoid DCs directly from peripheral blood.96
Naturally occurring DCs have shown promising results, and have indicated different efficacy with different DC subsets.96 For example, identifying whether the immunosuppressive environment of the tumour consists of regulatory T cells (Tregs) or tumour-associated macrophages may help in selecting the optimal DC subset (in this case conventional DC type 1 or 2, respectively) to induce the appropriate T-cell skewing.96 Similarly, modelling studies assessing the immune interactions of neo antigens in specific tumours have shown that neoantigens may act as a predictor of response to immunotherapy.97 As such, neoantigens may be useful in developing novel immunotherapies and personalised neoantigen-targeting vaccines.97,98
Summary and concluding remarks
The manufacture of DC vaccines is a complex process involving a number of technical challenges, including achieving the required yield for clinical use with appropriate purity, avoiding contamination by other blood components (e.g. platelets and RBCs), efficient maturation and antigen loading to produce viable mature DCs, and optimal administration of the vaccine to induce immunological responses in most patents. In addition, variation of MNC recruitment in donors has also been reported, though this requires further investigation.99
In recent years, the methods of manufacture have evolved and have started to meet these challenges, as well as improving convenience for the donor by reducing procedure time and product volume.45 The approaches for antigen loading have broadened, novel maturation signals have been developed and different sites of administration have been evaluated. As a result, robust, validated and GMP-compliant protocols have been developed for DC production.
Further development of these protocols is needed to produce standardised procedures for the preparation of cancer vaccines. Additionally, further refinements in vaccine strategies are needed to develop this promising therapeutic modality into a clinically meaningful treatment. This will facilitate larger multicentre clinical trials, with the aim of achieving widespread clinical adoption of therapeutic DC-based vaccines and potentially improved outcomes in a range of different cancer types.