Multiple myeloma (MM) is a malignant clonal disorder of plasma cells in the bone marrow, and more than 32,000 new cases are expected in the USA each year.1 Many clinical advances, including novel drugs and continuous therapy with treatment sequencing, have improved survival statistics.2 Despite this, MM remains largely incurable and the relapsed/refractory population remains vulnerable despite numerous new therapeutic options.3,4 Chimeric antigen receptor T (CAR-T) cell therapy offers early impressive results in the relapsed/refractory setting, providing hope that these therapies and others like it may be heralding in a new era in the treatment of MM, which may continue to move us closer to that elusive cure. CAR-T cell studies focus on the heavily pretreated patient with MM, and future studies that focus on its placement in the treatment sequence as well as optimum targets.
Background on CAR-T cells
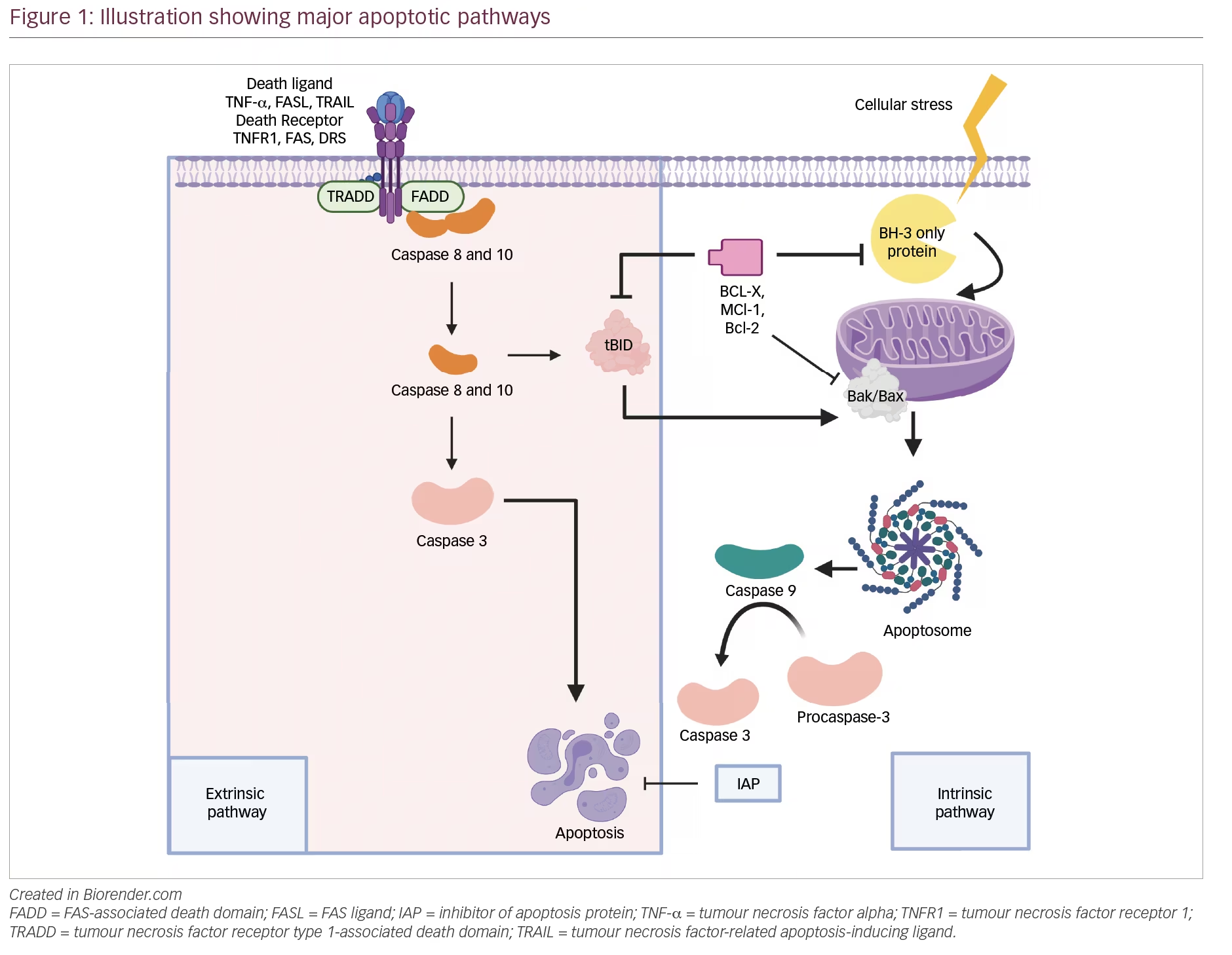
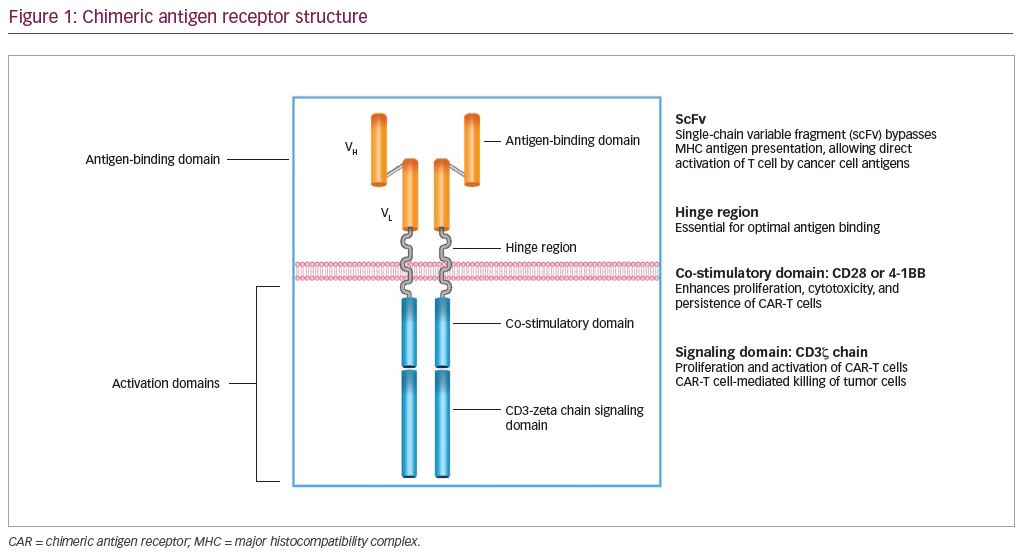
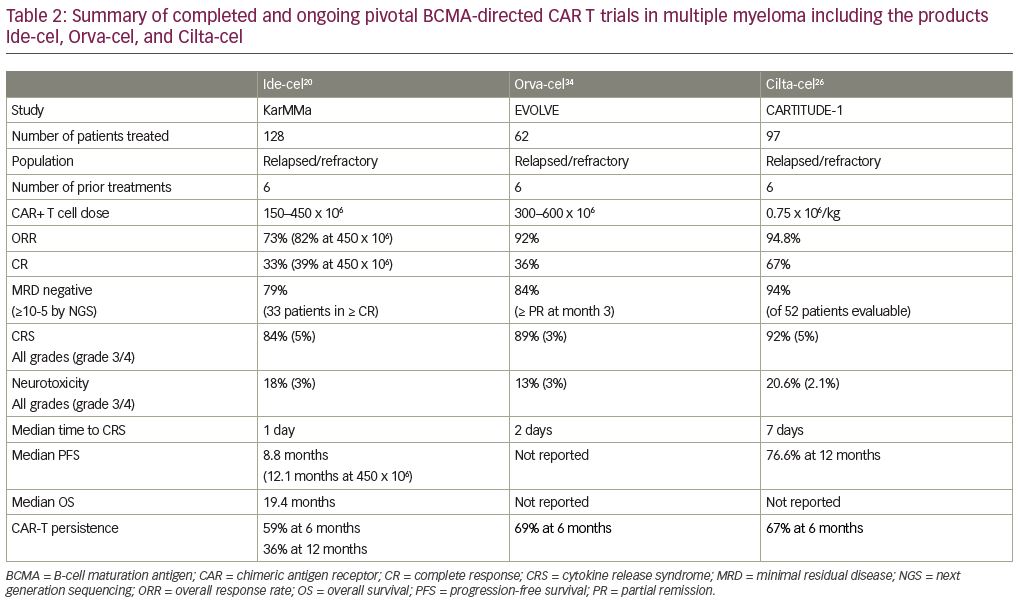
CAR-T cell therapy combines optimal antigen binding of a tumor cell surface molecule and direct T-cell activation inciting a targeted immune response as a novel class of “living” drug in malignancies. T cells are collected via apheresis and then genetically engineered to express an artificial receptor or CAR. The CAR consists of an extracellular antigen recognition domain or targeting domain, a transmembrane domain, and an intracellular domain to activate effector functions in the T cell.5 Second- and third-generation CARs combine additional co-stimulatory ligands, such as CD28 or 4-1BB signaling domains, to induce cytokine production and enhance function, differentiation, and persistence of the adoptive T cell (Figure 1).6
Multiple investigators concurrently working on cell therapy described a murine B-cell malignancy model in which T cells were transduced via a retroviral mechanism to target CD19.7 Treatment moved on to human subjects with acute lymphoblastic leukemia (ALL) and advanced B-cell malignancies. A phase II study using the anti-CD19 CAR-T product tisagenlecleucel in pediatric and young adult patients with CD19+ relapsed. Refractory B-cell ALL resulted in an overall remission at 3 months of 81% leading to the product’s US Food and Drug Administration (FDA) approval in August 2017.8 In advanced B-cell malignancies, a lympho-depleting conditioning regimen with fludarabine and cyclophosphamide followed by adoptive T-cell transfer in 22 patients showed an overall response rate (ORR) of 75% including 55% complete remission (CR) and 18% partial remission (PR).9 This was quickly followed with the FDA approval of axicabtagene ciloleucel in October 2017 after a phase II trial in 111 patients showed a 99% manufacturing success rate and an ORR of 82%.10
Multiple myeloma B-cell maturation antigen CAR-T target
Anti-CD19 CAR-T cell has been explored in MM in addition to aggressive B-cell malignancies. Although MM cells express CD19 infrequently, CTL019 cells are cytotoxic at low levels of CD19 expression. At the University of Pennsylvania, Philadelphia, PA, USA, a compassionate use pilot study of CTL019 cells were infused day 12 after high-dose melphalan and autologous stem cell infusion. The patient did not develop cytokine release syndrome (CRS) and obtained a CR.10,11 Despite this proof of concept, the majority of MM cells do not express CD19.
Since then there has been an explosion of CAR-T cell therapies in MM. B cell maturation antigen (BCMA) has been the most important target to date.12 BCMA—a type III transmembrane protein—is universally expressed on the surface of malignant plasma cells and a small fraction of mature B cells; however, it is not expressed on normal human tissue, including the primary human CD34+ hematopoietic cell. As a member of the tumor necrosis factor superfamily, its overexpression augments MM cell growth and survival serving as a receptor for B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL).13,14 BCMA maintains surface and intracellular expression through relapse, extramedullary spread, and residual disease after treatment, making it an attractive therapeutic target.15 Preclinical results of anti-BCMA CARs transduced with lentiviral vectors led to the specific killing of two-thirds of autologous primary MM cells leading to the initial in vivo studies.16 Other targets such as GPRC5D, SLAMF7, and CD138 are being developed as well, but are well behind BCMA.
First wave CAR-T cells for multiple myeloma
Initial clinical data
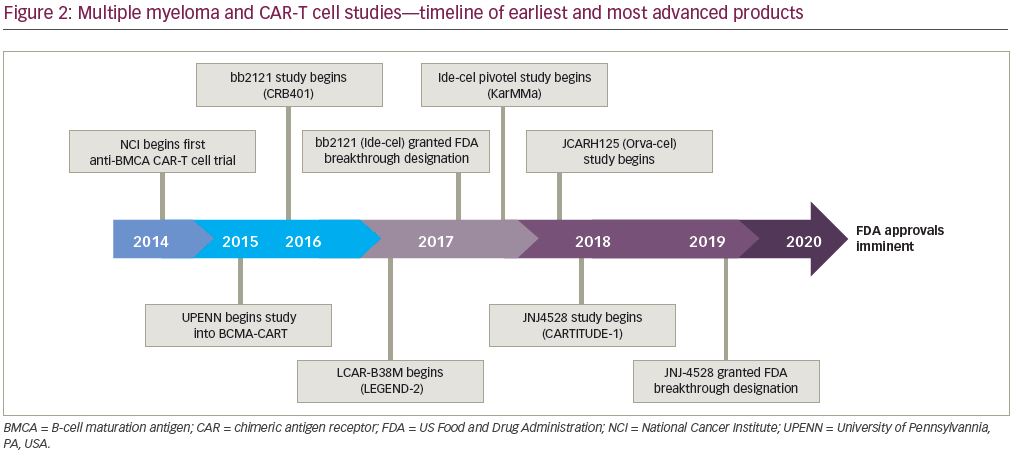
The first clinical data of a BCMA CAR-T cells included 12 patients with a median of seven prior treatments with an anti-BMCA CAR containing a murine single-chain variable fragment (scFv), a CD8α hinge and transmembrane region, a CD28 co-stimulatory domain, and a CD3ζ signaling domain. Patients were dosed from 0.3 × 106 CAR-T cells/kg up to 9 × 106 CAR-T cells/kg. This resulted in seven patients who achieved stable disease (SD): three with PR, one with very good partial remission (VGPR), and one with stringent complete remission (sCR). The two patients treated with the highest dose experienced complete and rapid elimination of bone marrow involvement from MM.17 In total, 26 patients were enrolled in this study with 16 at the highest dose level of 9 x 106/kg preceded by a conditioning regimen of cyclophosphamide and fludarabine.18 An impressive ORR of 81% was reported, and high peak blood CAR-T cell levels were reportedly associated with response. Importantly, all 11 patients who obtained remission greater than PR achieved minimal residual disease (MRD) negative status by flow cytometry. CRS, with hypotension, hypoxemia, fevers, and tachycardia, as well as coagulopathy and neurotoxicity with delirium, was described with the highest doses, which is similar to what has been seen in previous anti-CD19 CAR-T cell trials. Six of the 16 patients required vasopressors, but CRS was reversible in all cases. These positive results provided the foundation of future BCMA-directed CAR-T cell trials (Figure 2).
Ide-cel
Ide-cel (idecabtagene vicleucel, bb2121; Bristol-Myers Squibb, New York, NY, USA and bluebird bio, Inc., Cambridge, MA, USA) is a second-generation CAR-T cell product based on the initial product utilized by the Kochenderfer team. It is an autologous T-cell product transduced with a lentiviral vector encoding an anti-BCMA single-chain variable fragment, a CD137 (4-1BB) co-stimulatory domain, and a CD3-ζ signaling domain (Table 1). In the phase I CRB-401 study, ide-cel was infused using doses of 50 × 106 up to 800 × 106 CAR+ T cells. Responses were only seen at the 150 × 106 dose and higher, leading to 150 × 106 up to 450 × 106 CAR+ T cells in the expansion phase. The 33 enrolled patients were heavily pretreated with a median of seven lines of prior therapies. The most common ≥grade 3 toxicities were neutropenia (85%), leukopenia (58%), anemia (45%), and thrombocytopenia (45%). CRS was reported in 76% of patients, but were mostly grade 1 or 2, and only 6% were ≥grade 3. Neurotoxicity occurred in 42% of patients, and were mostly grade 1 or 2; only one patient (3%) had a reversible grade 4 neurotoxicity. The ORR was 85%, including 45% with complete responses—40% of whom subsequently relapsed. The median progression-free survival (PFS) was 11.8 months (95% confidence interval: 6.2–17.8). All patients who responded with ≥PR were evaluated for MRD-negative status (≤10−4 nucleated cells). Ide-cel expansion correlated with responses; interestingly, the responses were not BCMA-expression dependent. CAR-T cells subsisted up to 1 year after treatment.19
These findings lead to the confirmatory pivotal phase II KarMMa trial of ide-cel for patients with relapsed/refractory MM (RRMM) (Table 2).20 Patients who had ≥3 prior regimens (including immunomodulatory drug (IMiD), proteasome inhibitor (PI), and CD38 monoclonal antibody), and who were refractory to their last regimen as per International Myeloma Working Group (IMWG) criteria, were enrolled. After conditioning with cyclophosphamide and fludarabine, 150–450 × 106 CAR+ T cells were given to each patient. A total of 128 patients with a median age of 61 years received a median of six prior regimens; 84% were triple-refractory and 26% were penta-refractory. At a median follow up of 11.3 months, the ORR was 73% including 33% CR/sCR, with a median PFS of 8.6 months. The responses appear to be dose dependent; the 150 × 106 dose induced 50% ORR with each 25% CR and VGPR; the median duration of response (DOR) was not reached and the PFS was 2.8 months. The 300 × 106 dose appeared to be more efficient with an ORR of 69% including 29% CR, 14% VGPR and 26% PR; the median DOR was 9.9 months, and the PFS was 5.8 months. The 450 × 106 dose appeared to be the most efficacious with an ORR of 82% including 39% with CR/sCR, 26% with VGPR, and 17% with PR; the median DOR was 11.3 months and the PFS was 12.1 months. The median overall survival (OS) was impressive with 19.4 months for all patients and all subgroups, including older and high-risk patients, who appeared to benefit equally. In 33 patients who achieved CR/sCR, 79% were MRD-negative by next-generation sequencing (NGS) at a cutoff of 10-5 nucleated cells. Cytopenias (97%) and CRS (84%) were common, although CRS was mainly grade 1–2. The median onset to CRS was 1 day and only seven patients had grade 3–5 CRS. Neurotoxicity developed in 18%, but only 3% had grade 3 and no grade ≥4 were reported. A median peak CAR+ T cell expansion occurred at 11 days, and the expansion of CAR-T cells was higher in responders. CAR-T cells were detected in 59% of patients at 6 months and in 36% at 12 months post-treatment. An additional cohort enrolled three patients at an 800 × 106 dose with an ORR of 100%, including 100% with CR and MRD negative.21
Cilta-cel
LCAR-B38M is an autologous CAR-T cell product that also contains a 4-1BB co-stimulatory domain and a CD3-ζ T-cell signaling domain, and two BCMA-targeting single chain antibodies (Table 1). LEGEND-2 is a first-in-human, phase I, single arm, clinical trial conducted in RRMM performed at four centers in China using the LCAR-B38M construct.22 The study enrolled 74 patients at four sites with 57 patients enrolling from one study site. These 57 patients had a median of three prior lines of therapy; 68% were PI exposed, 86% IMiD exposed, and 60% had both prior PI and IMiD exposure. Few were daratumumab exposed. Patients received single-agent cyclophosphamide followed by three sequential infusions with LCAR-B38M at 20%, 30%, and 50% of the total dose. Common adverse events (AEs) reported were fever (91%), CRS (90%), thrombocytopenia (49%), and leukopenia (47%). Grade ≥3 AEs were reported in two-thirds of patients, specifically leukopenia (30%), thrombocytopenia (23%), and increased aspartate aminotransferase (21%). CRS occurred in 90% of patients and was primarily grade 1–2 (82%); only four patients (7%) had grade 3 events, and no grade 4–5 CRS was observed. Neurotoxicity was observed in only one patient (grade 1 aphasia, agitation, seizure-like activity). The median time to onset of CRS was 9 days, and all but one CRS events resolved. The ORR was 88%, and CR was 74%. Impressively, median PFS was 19.9 months and 28.2 months, respectively, for patients who achieved CR.23
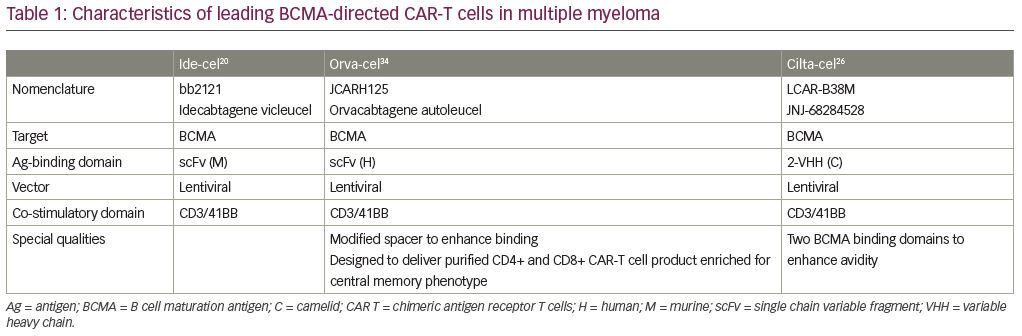
To confirm these results from LEGEND-2 in a more homogenous relapsed/refractory population, the phase IB/II dose-confirming study CARTITUTE-1 was initiated (Table 2). Cilta-cel (ciltacabtagene autoleucel, JNJ-4528, LCAR-B38M/JNJ-68284528; Janssen Pharmaceuticals, Beerse, Belgium and Legend Biotech Somerset, Ewing Township, NJ, USA) is identical to the CAR construct used in the LEGEND-2 study.24 In the phase Ib portion of the study, 29 patients received cyclophosphamide and fludarabine lymphodepletion and then a single infusion at a target dose of 0.75 x 106/kg. Patients were heavily pretreated with a median of five prior lines of therapy. Median time to CRS was 7 days, which is different when compared with the median onset of CRS from Ide-cel, which is only one day. Only 10% of patients developed neurotoxicity, and only 3% were ≥grade 3. The ORR was 100%, the sCR 86%, the VGPR ≥97%, and the PR 3%. The median time to sCR was 3 months, and the 9-month PFS was 86%. Of the 16 patients in CR, 13 were MRD-negative by NGS at a cutoff of 10−5 nucleated cells, and 11 at a cutoff of 10−6 nucleated cells. Interestingly, at the 6-month follow up, 22 of 28 patients had CAR+ T cells below the level of quantification in peripheral blood, suggesting that CAR-T persistence may not correlate with the depth of response to Cilta-cel.25 Updated phase Ib/II follow-up data in 97 patients with a 12.4-month follow up showed an ORR of 96.9%, and a CR of 67%. Median PFS was not reached and low-grade CRS occurred in 92% of patients, still with a later onset of 7 days. New neurotoxicity was 20.6% any-grade neurotoxicity, and 10.3% were grade 3 or greater. This was further divided into immune effector cell-associated neurotoxicity syndrome (ICANS) or other neurotoxicities, such as neurocognitive changes, nerve palsy, and peripheral motor neuropathy. ICANS of all grades occurred in 16.5% of cases, and ≥ grade 3 in 2.1%, with a median time to onset of 8 days and median duration of 4 days. Other neurotoxicities of any grade occurred in 12.4% of patients, and ≥grade 3 occurred in 9.3%, with a late median time-to-onset of 27 days and a duration of 75 days.26 Further phase II outcomes are eagerly awaited.
Mechanisms of resistance to B-cell maturation antigen CAR-T cell therapy
Despite impressive response rates and durability in RRMM patients, the majority will eventually relapse. The mechanisms of resistance to BCMA-directed CAR-T cell therapy are not well understood.27 Unlike CD19, where up to 75% of relapsed patients had loss of expression of the CD19 antigen on target cells, BCMA loss appears to be relatively uncommon.10,17 In a study by Ali et al., a partial loss of BCMA expression by malignant plasma cells was detected only in 1 of 12 patients. In the KarMMa trial only two patients who progressed were found to have lost BCMA expression on myeloma cells.17 This suggests that patients who progressed after BCMA-targeted therapy may very well respond to alternate BCMA-targeted therapy.28 It is imperative that these patients are not excluded from clinical trials directed at BCMA as is the current practice. Relapse appears to be occurring both in the setting of loss of CAR-T cells and in the setting of persistence of CAR-T cells. Thus various mechanisms appear to be at play including anti-BMCA CAR-directed antibodies and off-target binding of BCMA CARs on soluble BCMA (sBCMA). Additionally, reversible antigen loss through trogocytosis can occur by transferring the target antigen to T cells. This antigen loss leads to a decrease of target density on tumor cells, and inhibits T-cell activity via T-cell exhaustion or even T-cell killing. Furthermore, T-cell exhaustion with chronic antigen exposure, overexpression of programmed cell death protein-1 (PD-1) post infusion leading to functional BCMA CAR-T cell inhibition via PD-1/programmed death-ligand 1 (PD-L1) axis, and upregulation of tumor inhibitory signals have been postulated.17,29–32 As with prior therapies in MM, a combination approach seems to be necessary to best control the disease. Could the intrinsic heterogeneity of MM require that this same paradigm be followed with CAR-T cell therapy? Perhaps combinations with various immune-modulating therapies, such as IMiDs and anti-CD38 antibodies, may ultimately be required. This is also an opportunity where dual targeting CAR-T cell products may prove advantageous.
Second-wave CAR-T cells for multiple myeloma
Several new-generation products are now being tested clinically, building on the impressive results of the earlier products with the aim of overcoming some of the possible resistance mechanisms. The efficacy of gene transfer makes it feasible to express CARs in specific subsets of T cells. Central memory (Tscm) cells have superior proliferative capacity, longer telomeres, and improved survival after adoptive transfer compared to effector memory (TEM) and effector (TE) T cells.33 Thus, efforts to select for this T-cell phenotype has been a priority goal of the next generation of products.
Orva-cel
Orva-cel (orvacabtagene autoleucel, Juno Therapeutics, Seattle, WA, USA) is an anti-BMCA CAR containing a lentiviral construct with a fully human scFv, an optimized spacer, and 4-1BB co-stimulatory and CD3-ζ activation domains (Table 1). It avoids off-target binding with a low affinity for sBCMA, is active on target cells that express low BCMA density, minimizes tonic signaling to reduce antigen-independent exhaustion, and the modified spacer increases engagement of the antigen binding to the scFv, thus increasing cell kill.34 It is manufactured to produce an equal CD4:CD8 ratio enriched for a central memory phenotype thought to increase persistence and durability. The phase I/II EVOLVE trial enrolled 62 patients with RRMM who previously received a median of six prior lines of therapy. The trial included five dose level escalations of 50 x 106, 150 x 106, 300 x 106, 450 x 106, and 600 x 106 (Table 2). At the three highest dose levels, cytopenias were more frequent. Two deaths occurred due to infection and grade 5 mast cell activation syndrome (MAS)/hemophagocytic lymphohistiocytosis (HLH). Overall, CR occurred in 89% of patients with only two experiencing ≥grade 3 events, and had a median time-to-onset of 2 days, and a median time-to-resolution of 4 days. At a median follow up of 6.9 months, the ORR was 92%, with CR of 36%, VGPR of 32%, and PR of 24%. At the 600 x 106 dose level, 100% of patients were MRD-negative at 10-5 nucleated cells by NGS at 3 months. The trial has a 100% manufacturing success rate with robust expansion at all dose levels through day 29 with a trend towards increased expansion at greater dose levels, and 69% CAR persistence at 6 months. High baseline levels of sBCMA did not impact orva-cel activity. Based on these data, the phase II study plans to move forward at the 600 x 106 dose level.35
bb21217
bb21217 is identical to ide-cel (bb2121) utilizing the same scFv, 4-1BB co-stimulatory domain and CD3-ζ activation domains with the exception of co-culturing with a phosphoinositide 3 kinase inhibitor (PI3K) bb007 during ex vivo culture to enrich the drug product for T cells displaying a memory-like phenotype. In preclinical models, both CARs eliminated MM tumors in mouse models, but the bb21217 product was able to prevent tumor growth when re-challenged with a second tumor without reinfusion of cells. The bb21217 CAR showed higher levels of CCR7 and CD27, suggesting a higher level of memory-like T cells and lower levels of CD57, which is a marker of T cell exhaustion.36,37 In the phase I study, dose levels include 150 x 106 (n=12), 300 x 106 (n=14), and 450 x 106 (n=43). At a 23-month follow up of the first dosing cohort, the ORR is 83% and the median duration of response is 11.1 months.38 A proprietary manufacturing change took place at dose level 3. The original ORR was 60% with a CR of 32%. In the modified dose level 3 cohort, the ORR was 84% with a CR of 32%. All patients in CR were also MRD-negative, and the duration of response in this cohort at the 7-month follow up was not reached. Increased Tscm expansion correlated with the increase in level and the duration of response.39 In subsequent follow ups, it will be interesting if apparent longer persistence of the CAR-T cell product translates to more durable responses.
P-BCMA-101
P-BCMA-101 utilizes an anti-BCMA Centyrin™ (Aro Biotherapeutics Company, Philadelphia, PA, USA) fused to a CD3ζ/4-1BB signaling domain with a larger range of binding affinities, and is smaller, more stable and potentially less immunogenic. The piggyBac™ (PB) DNA modification system, as opposed to a viral vector, requires only plasmid DNA and mRNA producing an increased memory-like phenotype population. The higher cargo capacity allows for the incorporation of other genes, a safety switch that allows for rapid depletion of product in vivo if indicated by adverse events, and a selection gene that allows for enrichment of CAR+ cells.40 A phase I dose escalation study from 0.75–15 x 106 P-BCMA-101 CAR-T cells/kg was conducted in patients with RRMM; only one patient experienced low-grade CRS in the first cohort. Longer-term outcomes are still pending.41 The PRIME phase II study of the P-BMCA-101 product evaluates outpatient infusion as well as novel design features.42 A new manufacturing process utilizing a Nanoplasmid™ improved ORR at a dose of 0.75 x 106 from 66.7% down to 50% and will be utilized as the manufacturing process moving forward.43
CT053
CT053 is a second-generation CAR utilizing a fully human BCMA-specific single chain fragment variant (25C2) with high binding affinity. The CAR was initially studied in a single arm phase I investigator-initiated program in Eastern China. A median 17.7-month follow up reported on 21 patients receiving the 1.5 x 108/kg dose level after standard lymphodepleting chemotherapy (LDC). CR was low grade at 62.5% with a median onset of 3 days. Response showed an ORR of 87.5% with an impressive 79.3% CR rate. Median PFS was 18.8 months with improved DOR in patients without extramedullary disease (21.8 months versus 10.3 months). Persistence of the CAR-T cells was noted up to 161 days, and persistence correlated with response.44 To confirm these results, a phase Ib/II study, LUMMICAR-2, is enrolling subjects who have received three or more prior lines of therapy including a PI, an IMiD, and an anti-CD38 antibody.45 Patients receive standard LDC with a single infusion of CT053 with a dose level 0 targeted at 1.5–1.8 x 108/kg (n=8) and an escalated dose level 1 targeted at 2.5–3 x 108/kg (n=6). The manufacturing time was 8–10 days. Ten patients were evaluable at a median follow up of 4.5 months. CR rates were low grade 77% and 83%, with a median onset of 2 days and 4 days, at dose level 0 and dose level 1, respectively. An ORR of 94% with CAR-T cell expansion and persistence was noted for up to 6 months. The DOR and depth of response continues to deepen over time.45
Future of CAR-T cells for multiple myeloma
Incorporating CARs into earlier lines of therapy
While the second wave of CAR-T cell therapy in RRMM has focused mainly on product persistence to address the durability of response, future directions are multifaceted (Table 3). Both ide-cel and cilta-cel are in the process of conducting studies in the earlier relapsed setting of 1–3 prior treatments. Patients with high-risk MM are an attractive population in which to offer earlier CAR T-cell therapy given their significantly lower ORR, and shorter median PS and OS compared to standard-risk patients.
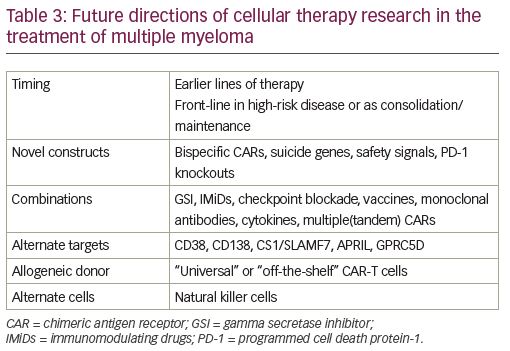
Bispecific CARs, CARs in tandem, novel targets
MM is a disease of high clonal competition and, as previously mentioned, antigen loss in a forthcoming clone can account for BCMA CAR-T cell relapse. Therefore, another approach to increase ORR and PFS in CAR-T cell therapy is dual antigen targeting to mitigate antigen loss with the treatment of different clones. Partnering BCMA targeting with anti-CD19 can trigger the elimination of malignant cells as CD19 is expressed on both myeloma cells and their progenitors. GC012F—a first-in-human BCMA/CD19 dual CAR—was constructed by linking BCMA and CD19 scFv joined by a CD8 hinge, transmembrane domain, co-stimulatory domain, and CD3 ζ. Patients have been enrolled at three dose levels with a primary end point of dose-limiting toxicity of GC012F, and secondary endpoints of MRD at the 3- and 6-month infusion, ORR, PFS, OS, and DOR. Sixteen patients have been enrolled, including six at the third dose level of 3 x 105/kg cells. The ORR of the study is 93.8% at 6 months, MRD-negative of 100% at 3 and 6 months at dose level 3, and no dose-limiting toxicity (DLT). Low CR grades are present at 87.5% with a median onset of 6 days.46
Other researchers have looked at using unique antigen targeting CAR-T cellss in tandem dosing to address higher-risk populations. A single-center, single-arm, phase II, feasibility study enrolled 21 patients who received lymphodepleting chemotherapy followed by humanized anti-CD19 CAR-T cells on day 0, and split dose murine anti-BCMA CAR-T cells on days 1 and 2. At a median follow up of 179 days, 20 of the 21 patients showed a response to treatment.47 Subsequently, a combined infusion has been escalated to 10 patients with R-ISS III disease with tandem autologous transplantation and combined infusion of CAR-T-19 and CART-BCMA cells post-transplant as consolidation treatment. CR occurred in all patients, limited to grade 1 and 2, and 100% of patients achieved VGRP at 100 days post autologous stem cell transplant (ASCT).48 Another trial is treating high-risk patients to consolidate their first or second line of therapy with CART-BCMA plus or minus huCART19.21 Non-BCMA targets—other than CD19—are also being investigated including products directed at CD38, CD138, CS1/SLAMF7, APRIL, and GPRC5D (Table 3).49–54
Gamma secretase inhibitors
A novel CAR product seeks to prevent MM relapse post CAR-T cell infusion utilizing GSI drug infusions with an anti-BCMA CAR. GSIs increase BCMA surface density and decrease sBMCA, thus theoretically increasing the efficacy of the anti-BMCA CARs. Patients received GSI (JSMD194) monotherapy administered every 48 hours over 5 days multiplied by three doses prior to lymphodepleting chemotherapy, and then infusion of a fully humanized BCMA CAR in combination with JSMD194 dosed three times weekly for 3 weeks, starting on the day of CAR infusion. The run-in dosing of JSMD194 increased plasma cell BCMA expression from 75% to 99%, and soluble BCMA decreased two-fold. BCMA antigen binding capacity increased to a median of 20-fold and best overall response rate was 100% in six evaluable patients.51
Allogeneic CAR-T cells
Until now, the majority of CAR-T cell products have been autologous. There are several issues that arise as a result, including: insufficient collection of T cells; failure of engineering BCMA CAR-T cells; suboptimal T-cell substrate; and a prolonged time period between collection and infusion of cells. Allogeneic CAR-T cells may be able to overcome these issues by providing readily available off-the-shelf CAR-T cells without the need for prior autologous collection. New issues introduced by the use of an allogeneic product is the potential for graft-versus-host disease (GVHD), and lack of persistence. There are several products currently in development and in early clinical trials. First studies are now open to enrollment evaluating the safety of allogeneic CAR-T cell products targeting myeloma cells. UCARTCS1 targets the antigen CS1, whereas the products BCMA-UCART, ALLO-715, CTX120, and PBCAR269A are all allogeneic BCMA CAR-T cell products that are currently being tested in phase I trials.49,50,55–57 Anti-BCMA ALLO-715 is an off-the-shelf CAR-T cell with 4-1BB co-stimulatory and CD3-ζ activation domains. The T-cell receptor alpha constant gene is disrupted to decrease GVHD risk, and the CD52 gene is disrupted to use ALLO-647 (an anti-CD52 monoclonal antibody) for selective and prolonged host lymphodepletion. The primary endpoint was tolerability with 31 patients with an average of five prior lines of therapy with all patients refractory to the last line of therapy; 94% penta refractory patients showed low CR rates (45%). Response and durability of the product were dose dependent, and at the third dose level (3.2 x 106 cells) the ORR was 60% with the VGPR at 40% or better.58
Natural killer CAR T cells
Natural killer (NK) cells can identify and kill malignant cells in the absence of antibodies and major histocompatibility complex molecules. CAR-NK cells are commonly engineered from donated umbilical cord blood NK cell lines, such as NK-92 or induced pluripotent stem cells.59 One of the possible concerns is the short in vivo persistence, though it is unclear if persistence of a CAR-T cell product is really necessary to induce a sufficient response as seen in the CARTITUDE-1 study (ClinicalTrials.gov Identifier: NCT03548207), where loss of persistence of CAR-T cells did not correlate with lack of response. Thus, it is quite possible that a brief and rapid onset of killing might be sufficient to induce a durable response. Compared to CAR-T cell therapy, the potential benefits of CAR-NK cell therapy include “off-the-shelf” availability, and a potentially low toxicity profile. Therefore, it might be advantageous to perform repeated and frequent dosing with these cell products similar to bispecific antibodies.60 Preclinical data show that genetic modification of NK-92MI cells with an anti-CD138 CAR enhances the cytotoxicity and potentiates the anti-myeloma effect with NK cell-based therapy. Both NKG2D-CAR NK cells and BCMA-CAR NK cells also appear to be equally efficient at eradicating MM cells.61 A phase I CAR-NK cell trial targeting BCMA for relapsed/refractory MM using adoptive BCMA CAR-NK 92 product is now open for enrollment.62
Conclusion
CAR-T cell therapy in the vulnerable RRMM patient population is showing unprecedented responses with single treatments of a biological agent as opposed to continuous therapy. Future directions are multifocal with increasing the quality of CAR products in terms of T-cell persistence with memory-like phenotypes, optimum CD4:CD8 ratios, and multi-antigen products, as well as timing of therapy in the treatment sequence. As patients progress on various treatments, they decrease their native T-cell populations, and have suboptimal T-cell input product for CAR-T cell production. Off-the-shelf CAR-T cell products from optimal T cell donors would avoid the problem of suboptimal autologous T cell collection and obviate the delays in CAR-T cell production as off-the-shelf cellular products are readily available. More provocatively, placing CAR-T cell therapy earlier in the sequence of treatment takes advantage of a more robust host T cell population for CAR-T cell production, and adds the possibility of potential cure. Additionally, future trials seek to increase CAR-T cell treatment efficacy with modifications such as bispecific targeting or novel combinations with IMIDs, monoclonal antibodies, and other anti-MM drugs. Moving CARs to the outpatient setting is also part of the next frontier of cellular therapy, demanding outpatient management and prevention of CRS. Despite advances in treatment, there are still socioeconomic barriers to treatment for advanced therapies for RRMM. Future treatment will depend not only on the CAR-T cell product manufacturing success and treatment response, but also on patient access.