Multiple myeloma is a plasma cell malignancy that typically develops in individuals in their late 60s with an average survival time of ~8 years.1 Despite recent advances in treatment, multiple myeloma remains largely incurable due to development of drug resistance in tumour cells. Patients often relapse even after autologous stem cell transplant and multiple lines of chemotherapy, and once refractory to current treatments, have a poor prognosis.
Multiple myeloma arises as a result of genetic changes during differentiation of B cells into plasma cell, which confer resistance to activation of the intrinsic apoptosis pathway.2 Apoptosis plays a key role in protection against genomic instability and maintaining tissue homeostasis and shapes the humoral immune response. This review article aims to understand the dysregulation of apoptotic pathways in multiple myeloma as well as its potential for targeted therapy.
Overview of the apoptosis
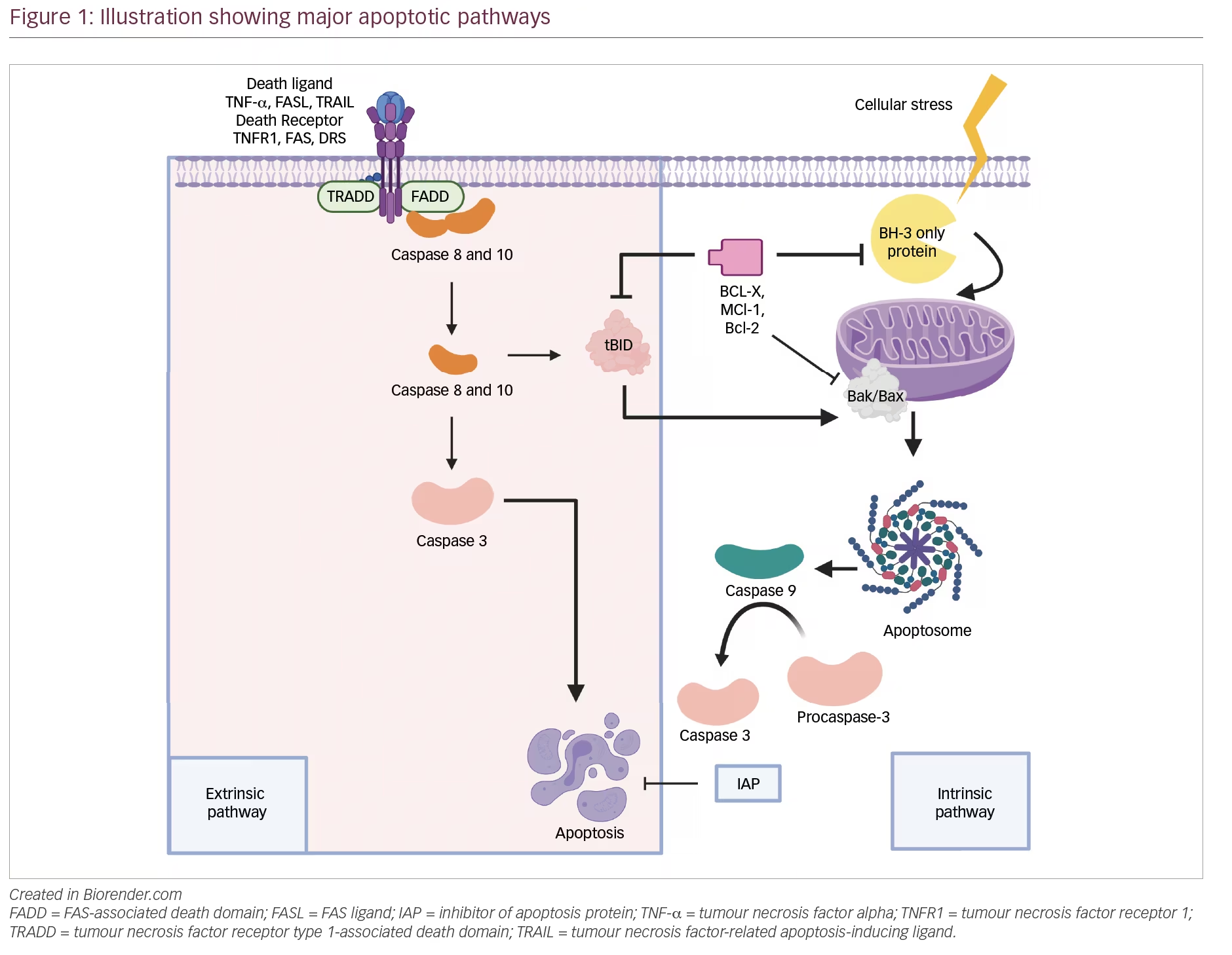
Apoptosis, also known as type I programmed cell death,3 is a crucial process for tissue homeostasis, characterized morphologically by cell shrinkage, membrane blebbing, chromatin condensation and nuclear fragmentation (Figure 1). It is triggered in response to cellular stresses like DNA damage, γ-irradiation, oncogene activation and growth factor withdrawal, and results in targeted elimination of damaged cells.4 The molecular web of apoptosis is broadly regulated by the following three proteins:3,5
- BCL-2 family of proteins: gatekeepers of death, involved in intrinsic pathway
- p53: the guardian of the genome
- The CD95 death receptor (also known as first apoptosis signal [FAS] or Apo-1), involved in extrinsic pathway.
BCL-2 family of proteins
BCL-2 was initially identified at the site of the t(14;18) chromosomal translocation in patients with B-cell lymphoma.6 This unique positioning of BCL-2 altered its transcriptional regulation, but this did not drive proliferation like other oncogenes. This led to identification of nearly 20 members of the BCL-2 protein family that drive oncogenesis by inhibiting programmed cell death. These members were grouped as anti-apoptotic (BCL-2, BCL-XL, BCL-W, MCL-1 and Bfl-1/A1) and pro-apoptotic (BAX, BAK, BOK, BAD, BIK, BIM, BID, PUMA and NOXA) based on sharing of at least one homologous domain (BH domains).7,8 These proteins appear to be the initial sensors (BH3-only proteins), regulators (anti-apoptotic) and effectors (BAX/BAK) of the intrinsic apoptotic pathway.8
Intrinsic/mitochondrial cell death
Various apoptotic stimuli, like DNA damage, growth factor deprivation and oxidative stress, activate the BH-3 proteins which initiate the mitochondrial pathway/intrinsic pathway.9,10 The BH-3 proteins (BAD, BMF, BIK and HRK) exert an inhibitory effect on anti-apoptotic BCL-2 proteins.11 The activator BH-3 proteins (BID, BIM, PUMA and NOXA) stimulate BAK and BAX protein.12 This leads to accumulation of BH-3–exposed BAK and BAX monomers, which oligomerize in the outer mitochondrial membrane causing membrane disruption and mitochondrial DNA efflux (also known as mitochondrial outer membrane permeabilization [MOMP]).3,12 MOMP represents the defining event that irrevocably commits a cell to die.8 A toroidal pore is formed in the mitochondrial outer membrane, which allows the release of intermembrane proteins like cytochrome c, second mitochondria-derived activator of caspase (SMAC) and Omi.13 Cytochrome c forms an intracellular complex called apoptosome with procaspase 9 and apoptosis protease-activating factor 1 (APAF-1), which leads to the activation of caspase 9.8,10 Caspase 9 further activates caspase 3/7,3 which leads to the terminal event of apoptosis. SMAC and Omi inhibit inhibitor of apoptosis proteins (IAPs)8 and X-linked IAPs, respectively, which are endogenous inhibitors of caspase function.14
Interestingly, the same stress signals that trigger apoptosis also stimulate the expression and release of heat shock proteins (HSPs), which repress apoptosis through inhibition of pro-apoptotic factors, such as p53, BAX, BID, AKT, APAF-1, and other BCL-2 families.15
Dodging the bullet: Pro-survival BCL-2 proteins
The first line of protection is provided by anti-apoptotic BCL-2, BCL-XL, and MCL-1, which sequester activator BH-3s (truncated BID [tBID]/BIM/PUMA) to prevent the initiation of BAX and BAK activation.3,5,16 BCL-2 can only inhibit BAX but not BAK. BCL-XL has higher protein stability and inhibits both BAX and BAK. MCL-1 has a short half-life and provides short-term, rapidly inducible enhancement of cell viability.17 It is also a cell cycle regulator and inhibits cell cycle progression through the S phase, ensuring cell survival until DNA repair is completed.18,19 As the second line of defence, the anti-apoptotic BCL-2 members can also sequester BH-3–exposed BAX and BAK monomers to prevent the homo-oligomerization of BAX and BAK.12
p53 protein
The tumour suppressor 53 (TP53) gene has been mapped to chromosome 17p13 and is known to function as a transcriptional regulator influencing cell cycle arrest (p21Waf1/Cip1), DNA repair and apoptosis in response to DNA damage (activation of BAX, NOXA, FAS and p53-inducible genes).18,20,21 p53 is regulated by overexpression of JNK, h-catenin, calpain-1 and auto-regulatory loops involving MDM-2 and Pirh2. p53 is also negatively regulated by epigenetic factors, like microRNA (miR)-125b and miR-504.22,23 MDM2 is an E3 ligase and promotes nuclear and proteasomal degradation of the p53 protein through a ubiquitin-dependent pathway. In unstressed cells, p53 is highly unstable with a short half-life of <30 minutes owing to rapid MDM2 degradation. Cellular stresses lead to stabilization of p53 tumour suppressor by blocking its degradation.24
The CD95 death receptor pathway (also known as extrinsic pathway)
Death receptors are a part of the tumour necrosis factor (TNF)/nerve growth factor-receptor superfamily, characterized by an intracellular death domain, essential for transduction of the apoptotic signal.25 This sub-family includes CD95 (FAS/Apo-1), DR3, TNF-R1 and two TNF-related apoptosis-inducing ligand (TRAIL) receptors.26 These death receptors are activated by their natural ligands, also known as death ligands, which are members of the TNF family (FAS ligand, TNF-α, and Apo-2 ligand/TRAIL). Once activated, it leads to trimerization of CD95 receptors and sequential recruitment of adaptor proteins FADD/TRADD (FAS-associated death domain/TNF-R1–associated death domain) and pro-caspase 8; hence, forming a death-inducing signalling complex.3,25 Induction of apoptosis occurs by two ways: i) type I – activation of caspase 8 and 10 is followed by rapid cleavage of caspase 3 and 7, which execute apoptosis;27 ii) type II – BID, a substrate of caspase 8, is cleaved into tBID by low amounts of death-inducing signalling complex. tBID translocates to the mitochondria and recruits the intrinsic pathway by causing release of cytochrome c and formation of apoptosome.25 Death receptors can trigger apoptosis independently of p53 and, therefore, can prove to be useful therapeutic targets.28
Dysregulation of apoptotic pathways in multiple myeloma
Multiple myeloma is characterized by infiltration of plasma cells in the bone marrow. The bone marrow microenvironment comprises extracellular matrix proteins and cellular elements including haematopoietic stem cells, immune cells, erythrocytes, bone marrow stromal cells (BMSCs), bone marrow endothelial cells, etc. Multiple myeloma cells interact with BMSCs by two different mechanisms: i) direct cell-to-cell contact between adjacent cell types, resulting in partial inhibition of proliferation of multiple myeloma cells and accumulation in the G0/G1 phase; ii) indirect production of soluble factors, which most likely inhibits mitochondrial apoptosis.29 The ability to evade apoptosis in the context of the microenvironment is a key characteristic of myeloma cells.
Adhesion of multiple myeloma cells to BMSCs and extracellular matrix in bone marrow initiates cross talks, which play a critical role in pathogenesis, cell survival and drug resistance in multiple myeloma. It upregulates cytokine secretion from both multiple myeloma cells and BMSCs, like TNF-α, vascular endothelial growth factor (VEGF) and transforming growth factor-β, which promote angiogenesis and stimulate BMSCs to secrete interleukin-6 (IL-6) and insulin-like growth factor-1 (IGF-1).17 Consequently, major signalling pathways, like nuclear factor (NF)-κB, JAK2/STAT3 and the phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), are activated, which promote cell cycle progression, inhibit apoptosis and lead to the production of other factors that support the growth and survival of myeloma cells.30 IL-6 is a major growth factor for multiple myeloma cells, and also promotes cell survival. IL-6 upregulates MCL-1 but blocks the nuclear accumulation of apoptosis-inducing factor p53 and the expression of p53-target genes, such as pro-apoptotic BCL-2 family members BIM and BAX.31,32 Phosphorylation of IGF-1 causes downstream activation of MAPK and PI3K/AKT signalling pathways in multiple myeloma cells, which further mediate cell survival by NF-κB activation and above-mentioned IL-6 signalling.16 The anti-apoptotic phenotype of myeloma cells is promoted by the bone marrow microenvironment through several anti-apoptotic proteins and anti-apoptotic signalling cascades as discussed below.
Nuclear factor κB/inhibitory unit of NF-κB
NF-κB is attached to a class of protein inhibitors called inhibitory unit of NF-κB (IκB) in unstimulated cells. Cell stimulation induces IκB phosphorylation and its destruction by proteasome; thus, NF-κB is released. Subsequently, NF-κB is translocated into the nucleus where it modulates gene expression.33
Genetic lesions confer gain of positive or loss of negative regulators of NF-κB signalling.34 For example, bi-allelic deletion causing inactivation of cIAP1 and/or cIAP2 is associated with an activation of the non-canonical NF-kB pathway.34,35 Dysregulation of NF-κB signalling in multiple myeloma has been linked to interaction with TNF ligands (B cell-activating factor [BAFF] and a proliferation-inducing ligand, APRIL) in the bone marrow.36,37 In healthy plasma cells, BAFF has been reported to induce the anti-apoptotic proteins BCL-2, A1 and BCL-XL and to reduce the pro-apoptotic protein BAK.37 The serum levels of BAFF and APRIL are higher in patients with multiple myeloma compared with controls, and increases with advancing stage of disease. It correlates with poor prognosis and shorter progression-free survival.38,39
The NF-κB index is high in both healthy and transformed plasma cells, and is relatively higher in multiple myeloma tumours and multiple myeloma cell lines with mutations in the NF-κB pathway. In contrast, multiple myeloma cell lines without apparent mutations in the NF-κB pathway have a substantially lower NF-κB index.40 Therefore, mutations in the NF-κB pathway in multiple myeloma tumours presumably result in less dependence on, or probably independence from, extrinsic signals from the bone marrow microenvironment.
NF-κB is a master regulator of cancer cells and helps them escape immuno-surveillance and promote tumour growth and inflammation. It mediates: i) cell survival and growth by positive regulation of BAFF, which in turn, activates NF-κB, AKT and MAPK signalling pathways that further regulate expression of other survival factors, such as BCL-2;34 ii) osteoclastic activity by regulating expression of macrophage inflammatory protein-1α, a chemokine involved in stimulating osteoclasts and causing bone destruction typical for multiple myeloma;41 and iii) transcriptional regulation of angiogenic molecules like VEGF, angiopoietin-1, TNF-α, IL-6, monocyte chemotactic protein-1 and IL-8.34
PI3K/AKT pathway
PI3K/AKT pathway is a complex signalling pathway with numerous feedback loops and crosstalk with numerous other pathways.42 DEPTOR (DEP domain-containing mTOR-interacting protein), a positive regulator of the PI3K pathway, is commonly upregulated in myeloma, especially in those with MAF translocations.18,42 But, unlike the MAPK pathway, the PI3K pathway members are rarely mutated in myeloma.43 Furthermore, hemizygous deletions of phosphatase and tensin homologue (PTEN), a negative regulator of AKT, have been reported in 5–20% of patients with multiple myeloma and multiple myeloma cell lines.17,44,45 Myeloma growth factors induce constitutive activation of the PI3K/AKT pathway, which leads to downstream BAD phosphorylation46 and formation of BAD-(14-3-3) protein heterodimer. This prevents binding of BAD to BCL-2 proteins and promotes cell survival.47
The activation of PI3K leads to the recruitment of serine-threonine kinase AKT to the cellular membrane and this enables the phosphorylation of multiple downstream targets, including mTOR, with various functions. mTOR induces downstream phosphorylation of ribosomal protein S6 kinase (p70S6K) and phosphorylation of eukaryotic translation initiating factor 4E-binding protein (4EBP1), which are involved in the control of survival and cell proliferation in myeloma cells.42 Deregulation of the PI3K pathway is thought to be important in myeloma as phosphorylated AKT, an indicative marker of pathway activity, is observed in approximately 50% of cases.48 High basal level endoplasmic reticulum (ER) stress in multiple myeloma induces downstream targets of AKT pathways, like NF-kB and phosphorylation of MDM2, which inhibits nuclear localization and degradation of p53 with a related decrease in expression of p53 target genes, such as BAX.17 AKT activation also negatively regulates pro-apoptotic proteins, BAD; caspase 9; forkhead family members, which induce CD95L like FKHR, FKHRL-1 and AFX; and glycogen synthase kinase 3.25,44,49 This contributes to myeloma cell proliferation and drug resistance.
JAK2/STAT3 pathway
Several autocrine and paracrine loops (particularly IL-6) activate the Janus family of kinase (JAK) by binding to gp130, which induces downstream phosphorylation and dimerization of the cytoplasmic signal transducer, and activator of transcription (STAT)3.50,51 This dimer translocates to the nucleus and activates transcription of target genes implicated in cell proliferation, differentiation and apoptosis.52 The JAK/STAT3 pathway is negatively regulated by the suppressors of cytokine signalling, and the SH2-containing phosphatases but these proteins are epigenetically inactivated in multiple myeloma by promoter hypermethylation.53
Catlett-Falcone and colleagues, reported that constitutively activated STAT3 is expressed in U266 myeloma cells at high levels, induces BCL-XL expression and confers resistance to FAS-induced apoptosis in U266 cells.54 MCL-1, another anti-apoptotic member of the BCL-2 family, has been reported to be induced by IL-6 in a STAT3-dependent way.55 However, in vitro studies by Quintanilla-Martinez et al., demonstrated that expression of anti-apoptotic proteins (i.e. BCL-XL, BCL-2 and MCL-1), in most cases, was independent of the expression and activation of STAT3.51
Vascular endothelial growth factor
VEGF is a hypoxia-induced angiogenic molecule that mediates autocrine and paracrine growth of myeloma cells.56 It confers protection to myeloma cells against apoptosis by promoting IL-6 production and upregulating anti-apoptotic proteins (MCL-1, surviving and cIAP).19 A statistically significant negative association has also been observed between VEGF and TRAIL.57 VEGF is a stronger upregulator of MCL-1 compared with IL-6 in patients with multiple myeloma upregulation. Studies in myeloma cells of patients demonstrate that about half of patients with multiple myeloma at diagnosis, and more than three-quarters of patients with multiple myeloma at the time of relapse, overexpress MCL-1.58 The high expression of MCL-1 correlates with disease activity as observed in more than 80% of human myeloma cell lines.58
Expression of VEGF is repressed by miRs (miR-15a/16: non-coding RNA, located at chromosome 13q14) in the post-translational phase. However, these miRs are downregulated in multiple myeloma.59 miR-15a/16 cluster has also been implicated in promoting apoptosis in multiple myeloma by downregulating BCL-2 proteins.59 Several other oncogenic miRs have been identified, which regulate several antitumour mechanisms, like induction of apoptosis, and promote multiple myeloma cell growth. miR-17-92 cluster exerts its oncogenic activity by targeting the pro-apoptotic gene, BIM, and miR-29b negatively regulates MCL-1 expression. miR-34a, another example of tumour suppressor miR, which is underexpressed in multiple myeloma and regulates BCL-2, CDK6 and Notch-1.45
Heat shock proteins
HSPs are highly conserved proteins involved in regulation of protein homeostasis and cell survival. Production of large amounts of immunoglobulins in multiple myeloma results in elevated expression of HSPs for protein handling.60 HSP90 sustains proliferative signalling pathways like AKT (PI3K/AKT pathway), IL-6R (JAK/STAT pathway), IκB kinases (NF-κB pathway) and APAF-1 (apoptosis), and resists cell death.61 On the other hand, cytoplasmic HSP70 regulates the apoptotic pathway at multiple levels. It protects BCL-2 from proteasomal degradation and blocks mitochondrial translocation of BAX thereby, preventing cytochrome c release and binds APAF-1; hence preventing the recruitment of caspase 9 to the apoptosome.60
p53 in multiple myeloma
TP53 mutations are present in 5–6% of patients newly diagnosed with multiple myeloma and 21–26% in relapsed and/or refractory multiple myeloma patients.62 The presence of TP53 mutations or del(17p) in the plasma cells is a poor prognostic factor and portends aggressive disease phenotype, a greater degree of extramedullary disease and shortened survival.18,62 There is also overexpression of MDM2 in multiple myeloma, which results in rapid turnover of p53 and therefore constant low levels of p53.63 MDM4, a homologue of MDM2, located on chromosome 1q is also implicated in p53 inactivation. Amplification of 1q is an indicator of high-risk disease and has also been recognized as an independent and significant prognostic factor.24
Epigenetic regulators, like miR-25 and miR-30d, directly target the 3′-UTR of p53 mRNA and subsequently result in decreased p53 protein expression and anti-apoptotic phenotype. Hypermethylation of p14ARF (negative regulator of MDM2), and the promoter region of TP53 gene itself, has also been implicated in pathogenesis of multiple myeloma.64,65
Resistance to extrinsic pathway in multiple myeloma
Regarding the extrinsic apoptotic pathway, multiple myeloma cells exhibit an elevated expression of Apo2L/TRAIL receptors.66 TNF, secreted by BMSCs, engages the NF-κB–IKK2 complex and stimulates transcription of pro-survival factors, such as cIAP1 and cIAP2, and produces a TRAIL refractory state in myeloma cells.35 TRAIL-induced apoptosis in multiple myeloma cells is also possibly blocked by decoy receptors such as osteoprotegerin, and an elevated expression of CFLAR/FLIP.66–69 Decoy receptors have a truncated cytoplasmic death domain, rendering them unable to signal and compete instead of receptor binding to Apo2L.70,71 Even though FAS and FAS ligand are increased in multiple myeloma and reflect disease progression,72 multiple myeloma cells are immune to FAS-mediated apoptosis.73 They suppresses immune responses against malignant and non-malignant cells by eliminating target cells by effector T lymphocytes.74
Targeting apoptotic pathways for treating multiple myeloma
A deeper understanding of the molecular pathways regulating apoptosis in multiple myeloma provides an opportunity to explore innovative therapeutic targets for treating multiple myeloma. This is especially important for patients with relapsed myeloma, refractory to proteasome inhibitors and immunomodulatory drugs.75 Recent preclinical data, with new selective inhibitors of apoptotic pathways in multiple myeloma, show promising antitumour effects both in vitro and in vivo myeloma models, either alone or in combination with novel agents (Table 1).
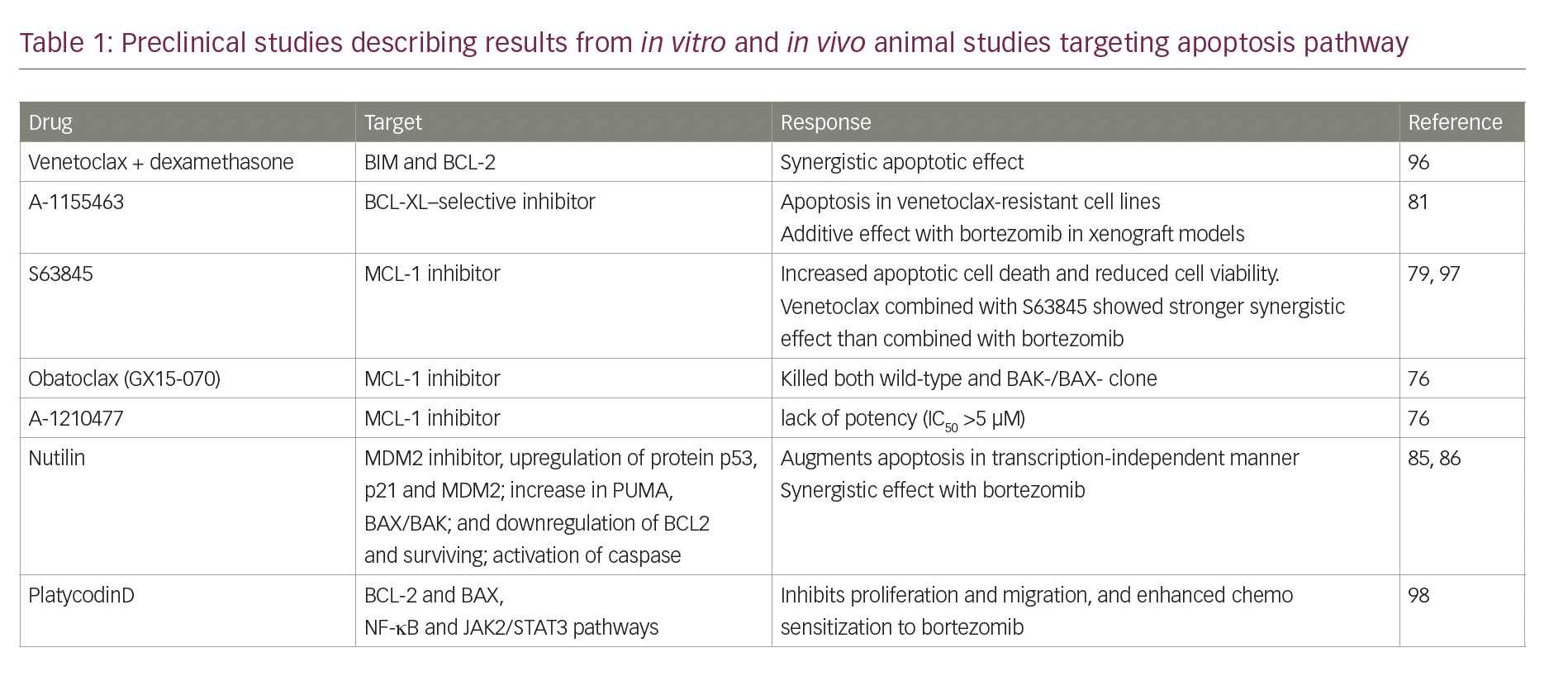
In vitro studies have shown that the majority of the myeloma cell lines rely mainly on MCL-1 followed by BCL-2 and BCL-XL for survival.76 In one study, targeting MCL-1 readily killed ∼70% of the myeloma cell lines tested, including both low-passage and well-established ones.76 Other in vitro studies have demonstrated that 17 of the 25 myeloma cell lines were highly sensitive to S63845 (MCL-1 inhibitor), not only in those with t(11;14), but also those with t(4;14) amplification of 1q or TP53 mutations, suggesting efficacy in cases refractory to standard-of-care agents.77,78 A synergistic effect was observed to be strong in doublet therapy with venetoclax + S63845, and even stronger with triplet regimen including S63845 + venetoclax and dexamethasone.79 Drugs targeting MCL-1 are in preclinical stages, and drugs that selectively inhibit BCL-2 and BCL-XL but not MCL-1, for example, ABT-737 and navitoclax (ABT-263), are undergoing clinical trials.80,81 The next-generation BH-3 mimetic venetoclax (ABT-199/GDC-0199) has shown robust pro-apoptotic effects in myeloma cell lines harbouring t(11;14) translocation; a cytogenetic subgroup expressing higher ratios of BCL-2 relative to MCL-1.81,82 Venetoclax resistance has been attributed to BCL-XL expression and can be mitigated by additive effects of bortezomib, which kills myeloma cells by upregulating NOXA and neutralizing MCL-1.81,83 These cell lines and xenografts were sensitive to a BCL-XL–selective inhibitor (A-1155463) as monotherapy and in combination with bortezomib.81
t(11;14) is considered neutral to prognosis, but del(17p) is universally associated with poor prognosis.84 Loss of p53 can make myeloma cells more resistant to cell death. Nutlin, one of the first potential inhibitors of p53–MDM2 interaction showed significant activity and synergy with standard-of-care drugs in cells with preserved p53 signalling pathway and wild-type p53.85,86 However, it was associated with resistance and clonal selection.87 Similarly, it was observed that RITA (reactivation of p53 and induction of tumour cell apoptosis), which binds directly to p53, induces cell cycle arrest and apoptosis in multiple myeloma cells resistant to other therapeutic approaches.88 Unlike nutlin, the efficacy of RITA was independent of the presence or absence of 17p deletion in primary patient samples, implying that RITA could be a therapeutic approach of choice in patients with TP53 abnormalities and drug resistance.89
Another approach to trigger apoptosis is restoring the original function of mutant p53 by small molecules called PRIMA-1 (p53 reactivation and induction of massive apoptosis), which could potentially prove beneficial in patients with p53 haploinsufficiency and total abolishment of p53 expression. In in vitro studies, Teoh et al. demonstrated a complete eradication of the multiple myeloma cells at 100 uM in all the human myeloma cell lines tested. PRIMA-1 exerts its cytotoxicity via UPR/ER stress pathway and might possibly also be a bortezomib re-sensitizing agent.90 APR-017 (PRIMA-1) and its methylated form APR-246 (PRIMA-1MET) are first-in-class drugs, currently in phase I/II clinical trials in patients with refractory haematological malignancies and prostate cancer.90 The cohort for this clinical trial by Lehmann et al. included only one patient with multiple myeloma. Hence, nothing concrete regarding its efficacy and dosing in patients with multiple myeloma can be said at the moment.91 However, the drug was well tolerated in the cohort, and the most common adverse effects were fatigue, dizziness, headache and confusion.91 Other novel approaches targeting the p53–MDM2 interaction with synthetic compounds, such as rocaglate or epigenetic regulators like miR-25 and miR-30d, have also shown promising results in human myeloma cell lines.85,92,93
Clinical studies targeting apoptotic pathways
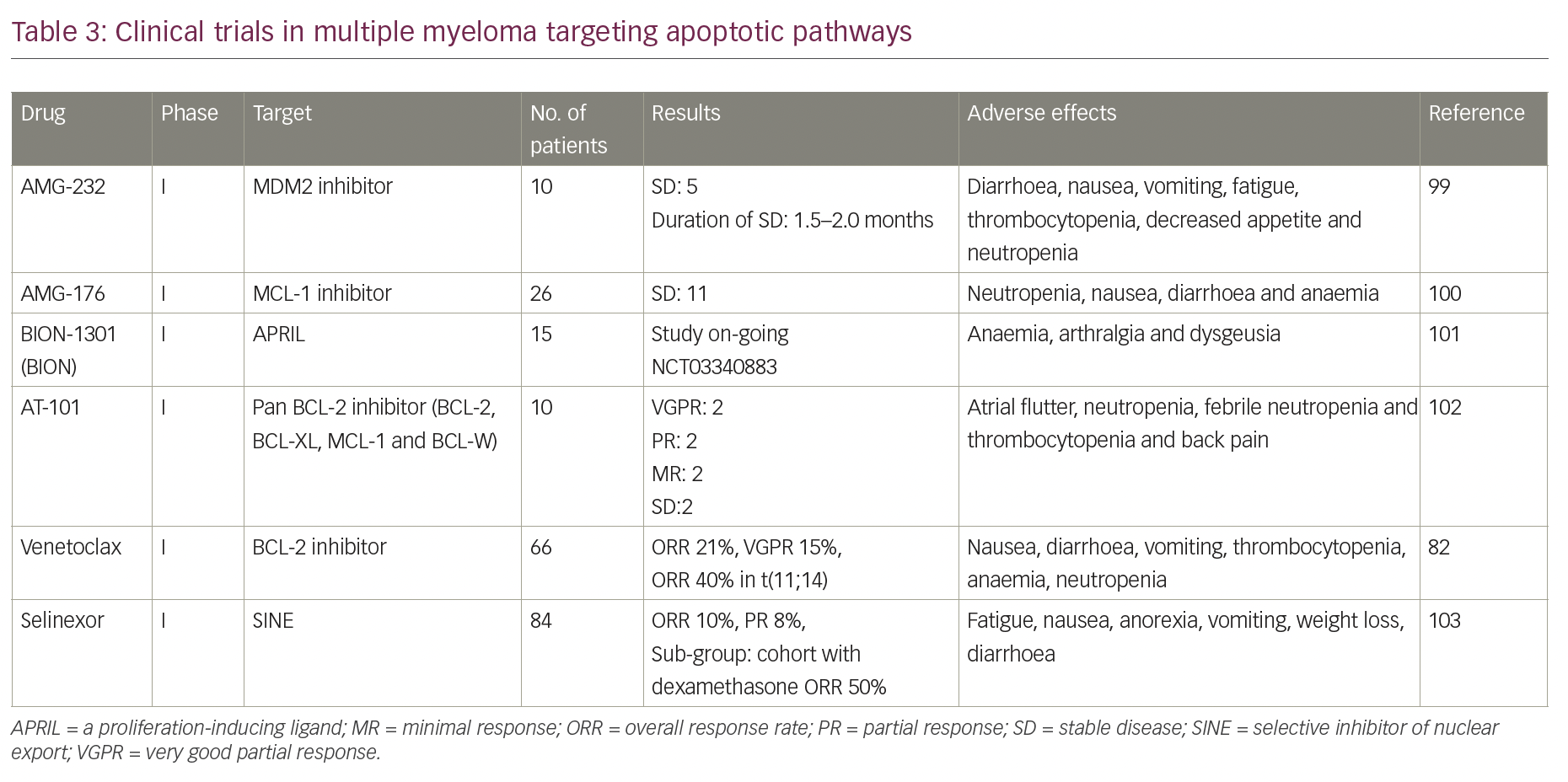
Establishing the function of apoptotic proteins in multiple myeloma, and their validation as therapeutic targets in myeloma cell lines and mouse xenograft models, can provide a framework for development of biologically-based treatments. Characterization of cytogenetic abnormalities in patients with multiple myeloma through fluorescence in situ hybridization and whole genome sequencing could also help design patient-specific targeted chemotherapy and predict response to therapy. Table 2 lists the on-going clinical studies targeting apoptotic proteins in multiple myeloma.
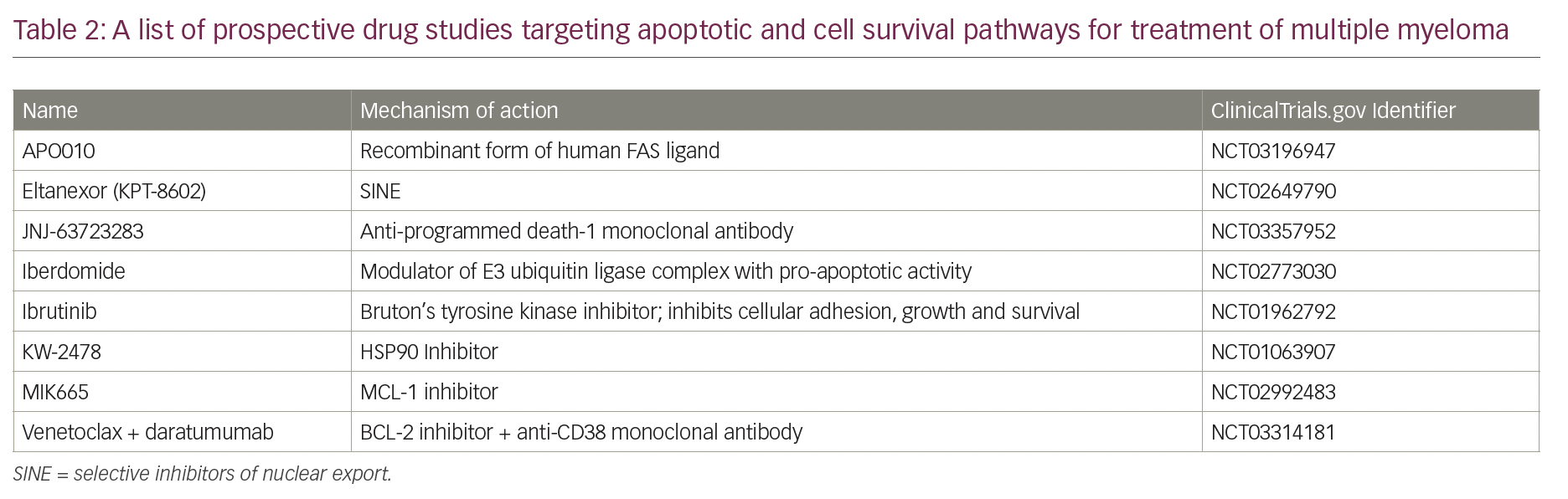
Although many drugs targeting apoptotic proteins are in phase I/II clinical trials (Table 3), venetoclax has shown the most promising results so far as monotherapy and in combination with standard-of-care drugs.82,83,94 The orally administered drug has an acceptable safety profile up to 1,200 mg and showed anti-myeloma activity in patients with relapsed/refractory multiple myeloma, predominantly in patients with t(11;14).82 In the study by Kumar et al., the overall response rate (ORR) in the entire cohort was 21%, and 15% had very good partial response (VGPR) or better.82 Among patients with t(11;14) and those with high expression of BCL2 protein, the response rates were substantially higher. Addition of dexamethasone to venetoclax increased the ORR to over 60% among patients with t(11;14) translocation. With the addition of bortezomib and dexamethasone to venetoclax, the ORR was 67%, with high responses among patients non-refractory to bortezomib who had received 1–3 prior lines of therapy (ORR, 97%; ≥VGPR, 74%).83 In another phase II dose-escalation study (ClinicalTrials.gov Identifier: NCT02899052), combining venetoclax with carfilzomib and dexamethasone in patients with relapsed, refractory multiple myeloma, the ORR was 97%, and 74% ≥VGPR, with an ORR of 100% in t(11;14) subset.94 However, in the phase III Bellini trial, which compared venetoclax with placebo in combination with bortezomib and dexamethasone, excess mortality was seen in the venetoclax arm, which was mostly related to infections in the context of disease progression.95
Conclusion
Programmed cell death is governed by a complex network of interactions between pro-survival (BCL-2, BCL-XL and MCL-1) and pro-death (BIM, BAD, BAK and BAX) BCL-2 family proteins. Overexpression of pro-survival proteins, especially MCL-1, has been observed to maintain survival in myeloma cells. Factors mediating myeloma cell growth, survival and the complex interaction of myeloma cells with the bone marrow microenvironment can serve as potential therapeutic agents, which may ultimately lead to improved disease‐free survival and potentially a cure for multiple myeloma.