The cachexia syndrome is seen across a wide range of chronic diseases and is especially evident in the cancer patient. This weight loss causes difficulties for the patient and clinician as it reduces quality of life (QoL) and also reduces tolerance of treatment. Weight loss is significantly associated with cancer morbidity and mortality.1,2 It is believed that up to 20 % of cancer patients die as a direct consequence of cachexia and that up to 50 % of cancer patients die with some degree of cachexia.3,4 The incidence of cachexia varies with tumour type, being lowest in sarcoma and breast cancers, whereas 80–90 % of pancreatic and gastric cancer patients experience weight loss.5
In cachexia, weight loss is attributed to a loss of both adipose tissue and skeletal muscle. However, it is the skeletal muscle wasting that likely contributes to excess morbidity and mortality in cancer patients.6 An international consensus recently defined cancer cachexia as a multifactorial syndrome characterised by an ongoing loss of skeletal muscle mass (with or without loss of fat mass) that cannot be fully reversed by conventional nutritional support and leads to progressive functional impairment. The pathophysiology is characterised by a negative protein and energy balance driven by a variable combination of reduced food intake and abnormal metabolism.7 A combination of primary and secondary anorexia, hyper-metabolism, hyper-catabolism and hypo-anabolism act together to aggravate weight loss.
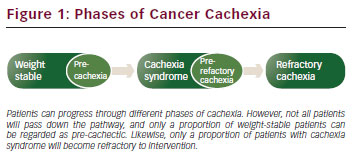
It is now recognised that cachexia exists through a range of phases (precachexia, cachexia syndrome and refractory cachexia) and it may be that even within the cachexia syndrome itself the more severe stages are less amenable to treatment and reversal. Clearly once a patient is confined to bed and moribund, the chances of reversal in muscle mass by multimodal rehabilitation are virtually gone. The greatest potential for prevention, recognition and reversal therefore lies with the precachectic state and during early cachexia syndrome.
According to international consensus, pre-cachexia may be recognised before any significant involuntary weight loss (i.e. >5 %) by clinical and metabolic signs such as anorexia and impaired glucose tolerance.8 Patients with >5 % loss of stable bodyweight over the past 6 months, or a body mass index (BMI) <20 kg/m2 and ongoing weight loss of more than 2 %, or sarcopenia and ongoing weight loss of more than 2 %, but have not entered the refractory stage, are classified as having cachexia.7 Refractory cachexia is characterised by a low performance status (World Health Organization [WHO] score 3 or 4) and a life expectancy of less than 3 months. This is an actively catabolic state where alleviation of symptoms is the mainstay of intervention.
Treating cachexia posses a difficult challenge to the cancer multidisciplinary team. The heterogeneity in presentation of cachexia has, in part, delayed formal descriptive terminology.
In addition, there is no validated classification of pre-cachexia. Moreover, although there are a range of biomarkers that might be used in this context (e.g. circulatory interleukin [IL]-6 or C-reactive protein [CRP] levels), the lack of longitudinal studies to integrate clinical classification with biological mechanism coupled with the complexity and duration of modern oncological management (chemotherapy, surgery, radiotherapy) makes this a difficult task.
Risk Stratification
Cancer cachexia is a continuum (pre-cachexia, cachexia syndrome, and refractory cachexia); however, not all patients traverse the entire spectrum. In fact it is plausible that some patients demonstrate protective mechanisms against progression. The risk of progression varies and depends on numerous clinical factors that should be taken into account when risk stratifying and counselling patients about weight loss in cancer and possible interventions.
Tumour type, tumour stage, co-morbidities, systemic inflammation, low food intake and response to anti-cancer therapy all play roles in determining whether a patient will be at risk of cachexia progression. Depending upon the tumour type, weight loss occurs in 30 to 80 % of cancer patients. Patients with pancreatic or gastric cancer have the highest frequency of weight loss, while patients with non-Hodgkin’s lymphoma, breast cancer, acute non-lymphocytic leukaemia and sarcomas have the lowest frequency of weight loss.9 Cross-sectional imaging analysis of skeletal muscle depletion is a powerful prognostic indicator. Utilising staging computed tomography (CT) scans and routinely collected patient clinical information, analysis of images can demonstrate previously occult muscle depletion that carries with it a poorer overall prognosis and likelihood to progress in cachexia severity.10 Measurements of systemic inflammation (Glasgow Prognostic Scale) have been shown to be reliable predictors of survival, independent of tumour stage, performance status, treatment (active or palliative) and has been shown in a variety of advanced common solid tumours.11 Knowing that cachexia is a metabolic process driven by the systemic inflammatory response the utilisation of such scores to risk stratify progression of cachexia seems intuitive. Clearly the most pertinent of these factors in risk stratification will be response to oncological treatment and therefore disease progression, for if the disease picture progresses the patient will almost certainly definitely succumb to weight loss. Moreover, as the clinical picture evolves through the treatment phase, the necessity to continually risk stratify and treat is heightened (see Figure 1)
A Complex Multidimensional Problem
The tumour’s role in the aetiology of cachexia includes the local secretion of pro-inflammatory cytokines (tumourkines) that initiate the host systemic inflammatory response/acute phase protein response (APPR),12 and the production of pro-cachectic factors that have direct catabolic effects on host tissues.13,14 Host mechanisms involve an aberrant response to the tumour’s presence, and include activation of both the APPR12 and the neuroendocrine stress response.15,16 The net result of such host–tumour interaction is an alteration in body composition, a major feature of which is a severe and specific loss of skeletal muscle mass.17
Besides the primary role of fat in storing excess lipids, adipose tissue is a major endocrine organ secreting hormones and cytokines (adipokines) that modulate appetite and nutrient metabolism. Therefore, alterations in adipose tissue mass can have significant effects on whole-body energy homeostasis.18 In the setting of cancer cachexia, there is emerging evidence that inflammatory signals from tumours intersect with the normal crosstalk between adipose tissue and other organs, leading to impaired energy balance and catabolism of fat and muscle.19
Although there may be loss of both fat and muscle tissue in cancer cachexia, it is the loss of skeletal muscle that will have the most profound effects on patients’ function and activities of daily living. In healthy adults, skeletal muscle mass is maintained by nutritional status and physical activity, reflecting a dynamic balance between protein synthesis and degradation. A predominance of either will result in muscle hypertrophy or atrophy. In cachexia, there is ongoing debate as to whether a reduction in protein synthesis, an increase in protein degradation or a combination of both is more relevant. Several functional studies have examined protein synthesis and protein degradation in rodent models of cancerassociated muscle wasting and revealed both reduced protein synthesis and increased protein degradation.20, 21
Inflammation and Signalling
Cachexia is an integrated physiological response of substrate mobilisation driven perhaps, mostly by inflammation. The consistent associations between inflammatory markers, such as circulating CRP and weight loss in cancer patients,22 reinforce the idea that cancer cachexia is linked to systemic inflammation that might be provoked by the spillover effects of excessive cytokines.
The tumour initiates a cytokine cascade that has multiple, direct, distant effects including the central induction of anorexia and the peripheral initiation of net skeletal muscle protein loss and the APPR.23
Cancer cells may rely on production of pro-inflammatory mediators for growth, protection from apoptosis and promotion of angiogenesis/ metastasis. A variety of human cancer cell lines have been shown to produce both pro-inflammatory and anti-inflammatory cytokines such as tumour necrosis α (TNF-α) and interferon γ (IFN-γ).24–27 TNF-α and IFN-γ work co-operatively to downregulate transcription of the myosin heavy chain (MyHC) gene in vitro and in vivo, but not other core myofibrillar proteins.28 TNF-α has been shown in vitro to inhibit both adipocyte and skeletal myocyte differentiation.29,30 It also plays a role in insulin resistance attenuating the insulin signalling pathway.31
Some cytokines may be potential repressors of cachexia. For example, IL-4, IL-10 and IL-13 all demonstrate anti-inflammatory, and hence presumably anti-cachectic, activity.32 Other cytokines (e.g. IL-15) may have potential ‘antioxidant’ properties that can counter the excess levels of reactive oxygen and nitrogen species.33,34 The final wasting status of the cachectic patient presumably depends on the balance between pro-inflammatory and anti-inflammatory cytokines.
Even with the same tumour type and burden, one individual may become cachectic whereas another will not. Such variation may relate to host genotype. Single nucleotide polymorphisms in the IL-1, IL-6 and IL-10 genes that are linked to production rates of these cytokines have been associated with the prevalence of cachexia in gastric or pancreatic cancer.35
The majority of known signalling pathways that contribute to muscle atrophy in pre-clinical models mediate their effects through activation of the ubiquitin proteasome pathway (UPP).36 Identification of up-regulation of two muscle-specific E3 ubiquitin ligases, MuRF-1 and MAFbx/atrogin-1, in at least 13 distinct animal models of atrophy,37,38 provides a compelling argument for a major contribution of the UPP in muscle wasting. Whether this is relevant to the clinical situation is still not clear. Moreover, although cytokines are known to activate the UPP pathway, the role of systemic versus intramuscular inflammatory signalling is poorly understood.
Myostatin/Activin
Myostatin, a TGF-b family member, is synthesised and secreted mainly from skeletal muscle cells and signals through the activin type II receptor, which then recruits an Alk family kinase, resulting in the activation of a Smad2 and Smad3 transcription factor complex.39 Overexpression of myostatin in mice leads to pronounced skeletal muscle atrophy,40 in the tumour-bearing state, although in animal models and patient muscle biopsy samples, evidence does indicate that myostatin levels and myostatin-associated signalling are activated as a result of a tumour burden.41
In addition to myostatin, there are other TGF-b family members induced by inflammatory cytokines. In particular, activin-A has been found to be upregulated in skeletal muscle after activation of the TNFa/TA K-1 signalling pathway.39 Furthermore, blockade of activin A in this model was sufficient to block atrophy. This finding demonstrates that, similar to the case of individual cytokines, blocking individual TGF-b family members such as myostatin alone may not be sufficient in settings of cancer cachexia.
Neural Control of Cancer Cachexia
The main molecular mechanisms regulating the cancer anorexiacachexia syndrome are alterations in brain neurochemistry, in particular, the hypothalamic melanocortin system with its activity diverted largely toward the promotion of catabolic stimuli leading to insulin resistance, increased lipolysis and accelerated muscle proteolysis.42 Proinflammatory cytokines (TNF-α and IL-1) and hypothalamic serotonergic neurons have been implicated in the dysfunction of the hypothalamic melanocortin system.43 Two peptide systems in particular appear to be strongly influential in the control of feeding behaviour: these are the orexigenic neuropeptide Y (NPY) and the anorexigenic proopiomelanocortin (POMC) systems.44 Many of these mediators exert their effects through induced changes in these two systems. They have been shown to be linked intricately with each other and to operate in parallel. POMC neurons are the source of the potent melanocortin neuropeptides, such as a-melanocyte-stimulating hormone which, via interaction with the central melanocortin receptors, induce an anorectic state. The role of cytokines in cancer anorexia may be affected through influence on both the NPY and POMC systems.45,46 These data strongly suggest proinflammatory cytokines such as IL-1 and TNF-α are involved in mediating the dysfunction of the melanocortin system leading to a catabolic state.
Human leptin is a protein of that is manufactured in and secreted primarily by adipocytes within white adipose tissue47 and is directly proportional to the total amount of fat in the body.48 Leptin is known to increase the frequency of action potentials in POMC neurons.49 In human cancer cachexia, leptin levels are markedly low,15 presumably because of weight loss, although other mechanisms may also be at play. Ghrelin, a 28 amino acid peptide produced by the P/D1 cells of the stomach, acts as the natural counterpart to leptin. Not only does ghrelin stimulate growth hormone (GH) secretion, but it also promotes food intake50 and decreases sympathetic nerve activity.51 Ghrelin has therefore been proposed as an alternative measure to GH therapy in muscle wasting. Patients with cancer cachexia demonstrate increased plasma concentrations of active ghrelin, suggesting a compensatory response to weight loss.52
Heterogeneity of Cachexia
Heterogeneity in all models of clinical and animal cachexia is one of the major factors that has impaired research into cancer cachexia. Rates of cachexia are influenced by tumour type, site and mass.5 Translation from a relatively acute setting of cachexia seen in animal models to the more chronic wasting seen in humans has been seen as a major stumbling block to develop therapies for human cancer cachexia. In humans, the cause of weight loss relates to not only the clinical status of the patient and specific effects of the tumour (e.g. causing bowel obstruction, tissue destruction or concomitant infection) but also to coexisting morbidities (e.g. heart failure, chronic renal failure or chronic obstructive pulmonary disease), age-related sarcopenia and the possibility of a genetic predisposition to develop cachexia.
The classical presentation of cachexia is of an extremely thin and wasted individual. However, heterogeneity in this clinical presentation is introduced by the current epidemic of obesity in the developed world. When healthy individuals develop a chronic disease, the higher risk associated with obesity is reversed and obesity becomes ‘protective’. This is known as the obesity paradox; however, this protective mechanism may not be afforded to cancer patients. Mean BMI of advanced cancer patients is now commonly measured at >25. There is, however, a subgroup of these overweight patients who hide gross muscle wasting under a mantle of adipose tissue. Approximately 40 % of overweight or obese patients with advanced pancreatic cancer have significant skeletal muscle wasting and this ‘myopenic or sarcopenic obesity’’ is an independent risk factor for accelerated demise.53 A recent survival analysis of 1,473 cancer patients highlighted the obesity/overweight epidemic in cancer patients with 52 % of this population being obese or overweight. Using CT cross-sectional imaging to visualise and quantify occult muscle depletion, they demonstrated that patients with cancer who are cachexic by the conventional criterion (involuntary weight loss) and by two additional criteria (muscle depletion and low muscle attenuation) share a poor prognosis, regardless of overall bodyweight.10
As skeletal muscle is a major target in cachexia, it is also relevant to consider heterogeneity as a result of sexual dimorphism. Men have more muscle mass than women and one might assume that this greater ‘reserve’ would be protective. However, weight loss and loss of muscle mass are greater in male than female cancer patients,54 and this may further relate to a high prevalence of hypogonadism in males.55
Treatment Options in Cancer Cachexia
Currently there is not an agreed protocol or guideline for the treatment of cancer cachexia. This problem is multifactorial including unsuitability of effective treatments due to side effects, failure of unimodal treatments at the clinical trial phase and a lack of therapies/clinical trials taking into account the multifactorial aetiology of cachexia. Unimodal therapies are less likely to achieve success against a complex metabolic process compared with a structured multimodal therapy tackling the interdependence of skeletal muscle, adipose tissue, the central nervous and immune systems combined. Future trials therefore require a stronger emphasis on combined treatment including nutritional support, appetite stimulation, exercise initiatives and multiple pharmacological treatments targeting this complex metabolism at several levels. The mainstay of treatment obviously remains eliminating the cancer; however, as this is often not possible. integration of nutrition, exercise and drugs must coincide with ongoing adjuvant cancer therapies.
Nutrition
Total energy expenditure is a combination of both the resting energy expenditure and the energy expenditure of physical activity. It is estimated that the cancer patient has a calorific deficit of roughly 200 Kcal/day.56 Protein intake should generally be maintained between 1–1.5 g/kg/ day.57 If these individuals were to participate in a diet that not only met basic nutritional needs but also have a net weight gain, they would need to increase calorific intake by 300–400 Kcal/day. This target becomes somewhat more difficult to achieve if the patient is already in the later stages of cachexia with significant anorexia and other co-morbidities. The most recent review of nutrition in malnourished cancer patients suffered from variability in the quality of studies and heterogeneity of endpoints.58 The findings suggested that oral nutritional interventions demonstrated no effect on survival and that the effect on bodyweight and energy intake was inconsistent but that there were some statistically significant improvements in some aspects of QOL. However it is not clear whether this represented clinically meaningful changes to the patient.
Drugs
No one class of drug has demonstrated any measurable advantage over the other in the treatment of cachexia. Either they have failed to demonstrate efficacy in clinical trials or they have an unwanted sideeffect profile. Many drugs have demonstrated promising results at the in vitro stage, however, have fallen down at the clinical trial phase. With increasing understanding of the metabolic interplay in cachexia, the rationale for therapy is trending towards agents designed to interfere with the inflammatory cascade and metabolic signalling.
Megestrol acetate (MA) is a synthetic progestogen agent first developed as an oral contraceptive in the 1960s. MA is currently used to improve appetite and to increase weight in cancer-associated anorexia. Although the mechanism is poorly understood its effect on appetite are thought to be exerted through an increase in the levels of NPY resulting in an attenuation of the firing of the ventromedial hypothalamic nucleus neurones, involved in the satiety mechanism.59 Other recent studies suggest that MA exerts effect on cytokines, which inhibit the action of TNF on fatty tissue and its products.60 In 2005 a systematic review was performed of MA for the treatment of anorexia-cachexia syndrome in patients with cancer, AIDS or other underlying pathologies. They concluded that MA improves appetite and weight gain in patients with cancer. However no advantage in QoL markers could be demonstrated due to the heterogeneity of data and endpoints.61
Corticosteroids have been shown to demonstrate an improvement in appetite and QoL. However due to their significant side-effect profile they should not be administered for long periods. Steroids are used extensively in routine oncology practice to counteract the effects of cytotoxic chemotherapy. Steroids are also used extensively to promote mood and appetite in the terminal stages of cancer.62
Inflammatory mediators contribute to the metabolic cascade and reduce appetite. Cyclooxygenase 2 (COX2) inhibitors reduce levels of acute phase reactants. N-3 fatty acids are also competitive antagonists of the inflammatory precursor arachidonic acid and can be given as fish oil capsules.63 A Cochrane review in 2007 concluded that the evidence was insufficient to give a recommendation. More recently small studies suggest that N-3 fatty acids may beneficially affect QoL, performance status and physical activity in patients with non-small-cell lung carcinoma (NSCLC) undergoing multimodality treatment.64,65
Thalidomide, which is thought to help reduce weight loss associated with cachexia in patients with cancer,66 modifies the cytokine triggers of the wasting response through its potent anti-TNF-α effects.67 A Cochrane review published in 2012 concluded due to the small number of studies and high heterogeneity among them, meta-analysis was not possible.68
IL-6 is a putative mediator of the systemic inflammatory response and muscle wasting. The humanised anti-IL-6 antibody BMS 945429 has been shown in pre-clinical phase I/II trials to reduce cachexia and improve fatigue of patients with NSCLC.69 In patients with cholangiocarcinoma, the administration of selumetinib (an inhibitor of mitogen-activated protein/extracellular signal-regulated kinase and of IL-6 secretion) demonstrated statistically significant mean muscle gain of 2.3 kg. The untreated group had a mean muscle loss of 1.2 kg. Neither group underwent assessment of QoL endpoints.70
Etanercept is a recombinant human TNF-α that specifically binds and renders soluble TNF-α biologically inactive by blocking their interaction with cell surface TNF-α receptors. In a recent phase I/II study of etanercept and gemcitabine, the only statistically significant outcome was a lowering IL-10 concentration.71
Two other studies have examined the role anti-TNF directed therapy in the treatment of pancreatic cancer and cachexia. One phase II study with thalidomide, an immunomodulator known to decrease TNF-α levels, demonstrated an improvement in weight and lean body mass at 8 weeks compared with placebo.66 By contrast, a phase II study of gemcitabine and infliximab, a monoclonal antibody blocking TNF-α, did not show a benefit in preserving lean body mass or survival.72
Despite a strong emphasis on targeting signalling pathways in cachexia, further study is necessary to demonstrate translational clinical benefit from in vitro studies to patient centred outcomes. Thus far, clinical trials targeting signalling pathways have struggled to demonstrate such an effect. Future studies targeting such pathways may consider utilising a multimodal approach to treatment and doing so at an early stage of disease/cachexia.
Anamorelin is a novel, oral mimetic of ghrelin.73 A randomised, doubleblind, placebo-controlled, crossover pilot study was conducted and demonstrated significant increase in GH, significant gains in weight and a significant improvement in patient-reported symptoms.74 Anamorelin is currently undergoing a phase III clinical trial (NCT 01395914)
GTx-024 (Enobosarm) is a non-steroidal selective androgen receptor modulator (SARM) that has tissue-selective anabolic effects in muscle and bone, while sparing other androgenic tissue related to hair growth in women and prostate effects in men. Enobosarm showed a dose-dependent improvement in total lean body mass and physical function and was well tolerated during a phase IIb trial of varied cancer types.75 Enobosarm is currently undergoing a phase III clinical trial for effects on cachexia in NSCLC (NCT01355484).
Myostatin is a member of the TGF-b family of secreted proteins, but unlike TGF-b, it is predominantly expressed in skeletal muscle (cardiac muscle and adipose tissue have low levels of myostatin), which plays a pivotal role in regulating skeletal muscle mass and function. Genetic evidence demonstrates this conclusively as deletion or knockout of the myostatin gene produces mice with a phenotype exhibiting a dramatic increase in the size and number of skeletal muscle fibres.76 Rodent models of cancer with cachexia clearly demonstrate raised levels of myostatin.77 Pharmacological blockade of the myostatin/activin-ActRIIB signalling pathway not only prevented further muscle wasting, but also reversed pre-existent loss of skeletal muscle. NCT 01505530 is a multicentre, randomised, double-blind, placebo-controlled trial in participants with locally advanced/inoperable or metastatic pancreatic cancer, and will investigate two doses of a myostatin monoclonal antibody (LY 2495655) in combination with chemotherapy. Its primary endpoint is survival however lean body mass and patient performance levels are secondary outcomes.
The acknowledgement that multiple components are responsible for the development of cachexia has led to the view that any cachexia intervention strategy should target all components i.e. multimodal therapy for a multimodal problem.78 Such strategies tend to target reversible clinical factors such as nutrition or exercise in combination with non-steroidal anti-inflammatory drugs or progestogens. Several recent studies conducting phase III trials have demonstrated favourable results that concluded that further multimodal studies are necessary.79,80
The critical concern remains when to start such treatment? Current multimodal anti-cancer therapy can take months or years, and patients frequently receive cytotoxic chemotherapy within 3 months of death. There is growing acceptance that anti-cachexia therapy cannot wait until such anti-cancer therapy is over (as has been the case with the majority of cachexia therapy trials in the past), but must form a major component of supportive oncology and be given along with potentially curative or palliative anti-cancer therapy.
Recent studies have emphasised that systemic cytotoxic chemotherapy can be associated with loss of skeletal muscle mass81 and it is potentially at this time, where muscle wasting may be both treatment and tumour induced, that cachexia therapy can have maximal impact. The results of several trials that aim to treat cachexia during chemotherapy are eagerly awaited.