The first primary microangiopathic haemolytic anaemia was identified as thrombotic thrombocytopenic purpura (TTP) by Dr Moschowitz in 1924. At that time, the classic pentad of clinical features, still used today, included fever, erythrocyte fragmentation, thrombocytopenia, kidney injury and neurologic injury.1 It is now known that this disease is characterized by the deficiency of a disintegrin and metalloproteinase with thrombospondin-1-like motifs, 13th member (ADAMTS13), a metalloproteinase that cleaves von Willebrand factor (VWF) multimers. In the normal state, VWF plasma levels are maintained by an endothelial cell (EC) constitutive pathway, although a specialized secretory pathway retains the capacity for exaggerated VWF release from EC-compartmentalized Weibel–Palade bodies. VWF released into the bloodstream following vascular damage serves as the bridge between EC-exposed subendothelial collagen and platelet glycoprotein Ib (GP1b), thereby regulating primary haemostasis through the first phase of platelet–endothelial adhesion (see Figure 1).
Figure 1: Pathophysiology of thrombotic thrombocytopenic purpura with medications explored in recent clinical trials
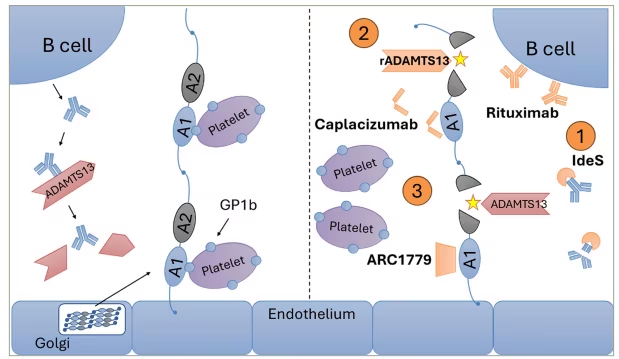
Left: VWF is biosynthesized and secreted from EC Golgi, resulting in extracellular high-molecular-weight VWF multimers that are proteolytically cleaved by ADAMTS13 (normal state). Shown are the critical VWF A1 and A2 domains, which mediate platelet GPIb/collagen interactions (A1) or serve as the site of ADAMTS13 cleavage (A2). In TTP, loss of ADAMTS13 protease activity (either in cTTP or iTTP) leads to loss of proteolytic cleavage and accumulation of EC-derived ultra-large VWF multimers (depicted here as A1 and A2 domains), followed by exaggerated platelet GB1b binding and uncontrolled platelet activation. Right: Clinical trials explore agents that (1) interfere with the production of B-cell-derived IgG antibodies (i.e. rituximab and IdeS), (2) replace ADAMTS13 using a recombinant product (rADAMTS13) or (3) inhibit VWF/platelet interactions by blocking VWF A1 domains (caplacizumab and ARC1779), which mediate platelet GP1b binding.
ADAMTS13 = a disintegrin and metalloproteinase with thrombospondin-1-like motifs, 13th member; cTTP = congenital thrombotic thrombocytopenic purpura; GP1b = glycoprotein Ib; IdeS = IgG-degrading enzyme of Streptococcus pyogenes; IgG = immunoglobulin G; iTTP = immune thrombotic thrombocytopenic purpura; rADAMTS13 = recombinant ADAMTS13; TTP = thrombotic thrombocytopenic purpura; VWF = von Willebrand factor.
VWF is originally synthesized in ECs and dimerized in the endoplasmic reticulum. Subsequently, VWF multimers assemble in the acidic conditions of the Golgi apparatus and are ultimately released as high-molecular-weight multimers, which are secreted from the EC. In contrast to VWF multimers released through the constitutive pathway, VWF is also stored in EC-specific Weibel–Palade bodies where its release causes unravelling into ultra-large strings that remain attached to ECs. These ultra-large multimers remain in an active conformation, and in physiological and stress pathways are subsequently cleaved by ADAMTS13, thereby reducing the size and functional activity of VWF for its platelet receptor.2
In the pathological state, a deficiency of the essential ADAMTS13 leads to the accumulation of ultra-large VWF multimers on endothelial surfaces, causing exaggerated and uncontrolled platelet aggregation. Subsequent formation of microthrombi in small vessels leads to ischaemic damage to end organs, including the central nervous system and kidneys. Platelets are consumed, leading to thrombocytopenia. Finally, damage to erythrocytes travelling through occluded small vessels leads to morphologic disruption and classic schistocytes seen with microangiopathic haemolytic anaemias.
TTP is now recognized to consist of a classical acquired form, mediated by an autoantibody against ADAMTS13 (immune thrombotic thrombocytopenic purpura [iTTP]), and a congenital deficiency of ADAMTS13 (congenital thrombotic thrombocytopenic purpura [cTTP]), also known as Upshaw–Shulman syndrome. Treatment with plasma exchange (PEX) is now known to (1) replace ADAMTS13 protein and (2) remove circulating autoantibodies, with striking reduction in mortality rate to approximately 20%.3 However, relapses occur in 41–74% of patients.4,5 Given that 16% of cases are refractory to primary treatment with PEX and corticosteroids, ongoing efforts continue to identify and characterize new medications that may improve remission rates, prevent relapses and improve long-term outcomes.3
Diagnostic criteria and principles of management
Based on recent advances in pathophysiology and management, current guidelines provide the following criteria for diagnostic and clinical decision-making (Table 1).6–9 A clinical risk assessment model such as the PLASMIC score or the French score may be used to determine the pretest probability of TTP and guide treatment decisions.9 For example, the PLASMIC score uses the following criteria: platelet count less than 30 x 109/L; evidence of hemolysis (defined as indirect bilirubin >2.0 mg/dL, haptoglobin undetectable, or reticulocyte count >2.5%); no active cancer; no prior solid organ or stem cell transplant history; mean corpuscular volume (MCV) less than 90 fL; international normalized ratio (INR) less than 1.5; and serum creatinine less than 2.0 mg/dL.10 Cases fulfilling 5 criteria, or at least 6 criteria, indicate an intermediate and high pre-test probability of ADAMTS13 activity <10%, respectively. Plasma testing for ADAMTS13 activity and inhibitor should be sent prior to PEX or any blood product.11 PEX and steroids (either prednisone 1 mg per kilogram per day or pulse dose methylprednisolone) should be initiated once the diagnosis is suspected.6 In patients with high pre-test probability, caplacizumab may be considered before ADAMTS13 results are available.11 An ADAMTS13 activity of less than 10% (a positive result) confirms the diagnosis in patients with clinical suspicion. In patients with a diagnosis confirmed by ADAMTS13 activity <10%, rituximab should be added to primary therapy, and caplacizumab should be added if not already started.7,11 Rituximab is generally infused 18–24 hours apart from PEX to avoid removal of the drug via pheresis.6 In patients with ADAMTS13 activity 10–20% (an equivocal result), continuation of therapy is determined by clinical judgement. In patients with low or intermediate pretest probability on a risk assessment model or with ADAMTS13 10–20% or >20% (a negative result), alternative diagnoses such as haemolytic uraemic syndrome or disseminated intravascular coagulation should be considered.
Table 1: Summary of ASFA 2023 and ISTH 2025 treatment guidelines6–8
| Disease | Therapy | Recommendation type | Certainty of evidence | Society |
| iTTP, acute | Plasma exchange | Category I | 1A | ASFA6 |
|
| Corticosteroids | Strong | Very low | ISTH7 |
|
| Rituximab | Conditional | Very low | ISTH7 |
|
| Caplacizumab | Conditional | Moderate | ISTH7 |
| iTTP, remission and low ADAMTS13 activity | Rituximab | Conditional | Very low | ISTH7 |
| cTTP, remission | ADAMTS13, recombinant-krhn | Strong | Moderate | ISTH7,8 |
|
| Plasma infusion (10-15 mL/kg every 1–3 weeks) | Conditional | Very low | ISTH7,8 |
Summary of treatment recommendations for immune and congenital TTP. In the acute setting, the ISTH recommends the addition of rituximab to corticosteroids and PEX over corticosteroids and PEX alone. The ISTH further suggests using caplacizumab over not using caplacizumab. For patients with cTTP in remission, prophylaxis with recombinant ADAMTS13 is recommended. If recombinant ADAMTS13 is not available, the ISTH suggests prophylaxis with plasma infusion over a watch-and-wait strategy.
ADAMTS13 = a disintegrin and metalloproteinase with thrombospondin-1-like motifs, 13th member; ASFA = American Society for Apheresis; cTTP = congenital thrombotic thrombocytopenic purpura; ISTH = International Society on Thrombosis and Haemostasis; iTTP = immune thrombotic thrombocytopenic purpura; PEX = plasma exchange; TTP = thrombotic thrombocytopenic purpura.
Numerous adjuvant therapies in addition to PEX have been investigated in the past 15 years. This article expands on recent clinical trials in adjuvant therapies in TTP, including ongoing and terminated trials.
Aims
This manuscript intends to review clinical trials regarding adjuvant therapy in TTP in the past 15 years, as well as new medication approvals in TTP.
Learning objectives
Upon completion of this article, readers will be able to describe new therapies for TTP and their indications. Furthermore, readers may outline terminated trials and treatments proven to be ineffective.
Methods
A PubMed search was conducted with the phrase “(purpura, thrombotic thrombocytopenic[MeSH Terms]) AND ((“2010/01/01″[Date – Create] : “2025/03/29″[Date – Create]))”. A concurrent search was performed under the ClinicalTrials.gov registry for ‘TTP’ and ‘thrombotic thrombocytopenic purpura’ for interventional studies from 01 January 2010 to 29 March 2025. Only human clinical trials with investigational adjuvant agents for TTP were included. Trials evaluating PEX alone were excluded. Retrospective registry analyses were excluded.
One reviewer was used to screen each record and report. Outcomes of interest including time to platelet response, relapse rate, overall response rate and trial status were documented. Outcomes including IgG-specific endopeptidase exacerbations, defined as episodes of thrombocytopenia occurring within 30 days from last PEX, and relapses, defined as recurrences more than 30 days from last PEX, were also sought.12
Results
On PubMed, 35 records were identified using the above-mentioned search criteria. On the ClinicalTrials.gov registry, 13 studies were identified.13–25 From these studies, 15 records were identified and 9 were duplicated in the PubMed database. One study was excluded as it was related to myeloma and not TTP; 40 reports were thus assessed for eligibility.26
Three trials were excluded because they evaluated therapeutic PEX alone or differences in blood products used during PEX without additional therapy.27–29 Further reports were excluded as they did not describe clinical trials. As a result, two case reports were excluded.30,31 Two diagnostic studies were excluded.32,33 Four registry studies were excluded.34–37 Two observational studies were excluded.38,39 One retrospective study was excluded.40
Overall, 26 records of 21 clinical trials for adjuvant therapies were identified and included in the analysis. A flow diagram demonstrating the review process is included in Figure 2. The clinical trials are summarized in Tables 2–4.5,13,14,41–66 A timeline of select clinical trials and US Food and Drug Administration (FDA) approval dates is included in Figure 3.
Figure 2: Flow diagram depicting the review process
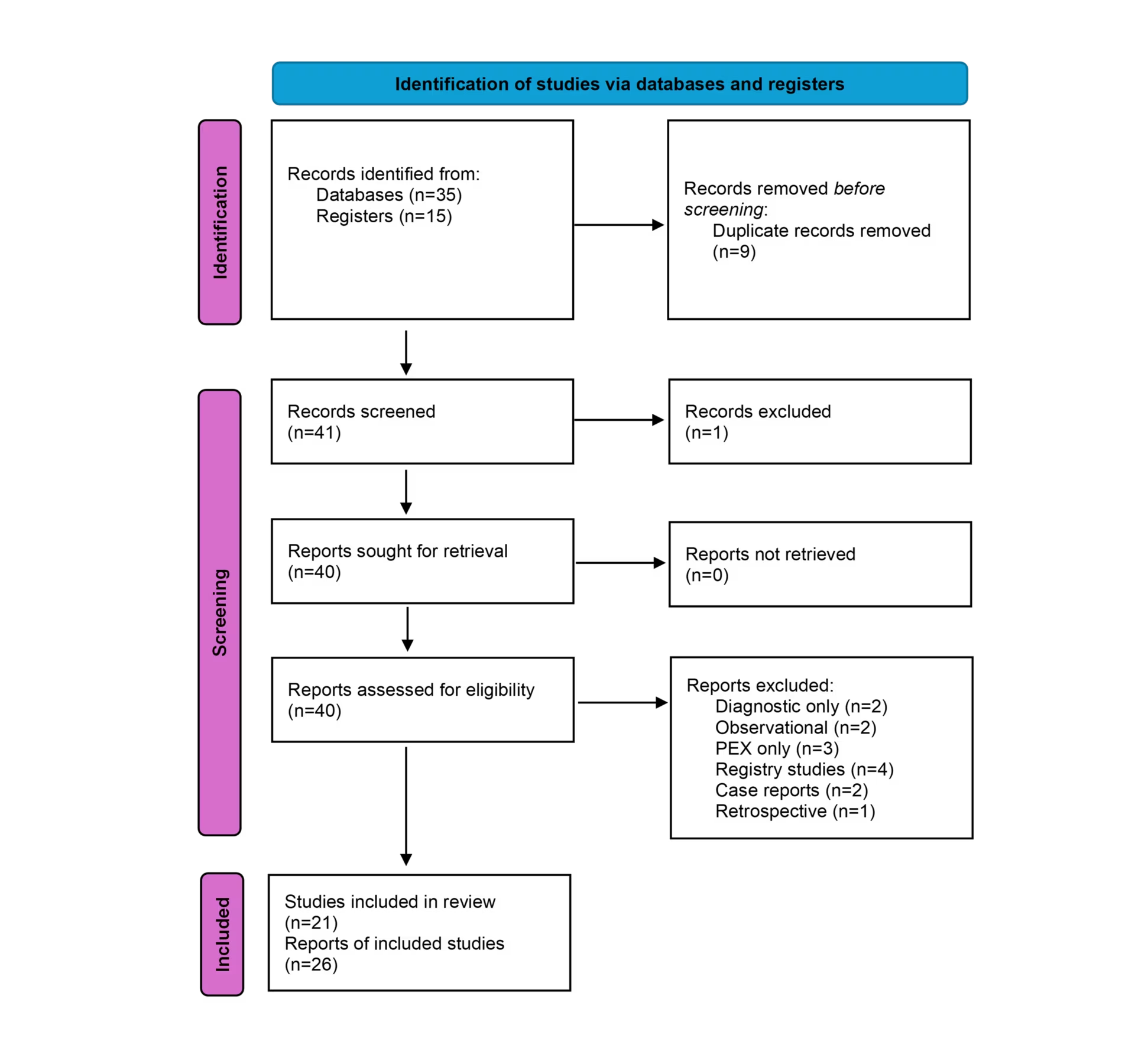
PEX = plasma exchange
Table 4: Trials in progress14,41
| # | Treatment | MOA | Ind | Status | Study design | Trial name, trial identifier, and phase |
| 1 | ADAMTS13, recombinant-krhn | rADAMTS13 | iTTP | Not yet recruiting | Randomized controlled trial, double blind rADAMTS13 with PEX versus rADAMTS13 without PEX | A Study of TAK-755 (rADAMTS13) With Little to No Plasma Exchange (PEX) Treatment in Adults With Immune-mediated Thrombotic Thrombocytopenic Purpura (iTTP)14 NCT05714969 Phase IIb |
| 2 | ADAMTS13 recombinant-krhn | rADAMTS13 | cTTP | Recruiting | Open-label single arm continuation study of patients in NCT03393975 | A Study of TAK-755 in Participants With Congenital Thrombotic Thrombocytopenic Purpura NCT0468300341 Phase IIIb |
Summary of clinical trials in progress for TTP as of April 2025.
cTTP = congenital thrombotic thrombocytopenic purpura; iTTP = immune thrombotic thrombocytopenic purpura; MOA = mechanism of action; PEX = plasma exchange; rADAMTS13 = recombinant ADAMTS13; TTP = thrombotic thrombocytopenic purpura.
Table 2: Clinical trials evaluating adjuvant therapies for immune thrombotic thrombocytopenic purpura5,13,42–63
| # | Treatment | MOA | N | Population | Study design | Trial name, trial identifier, year and phase |
| 1 | Caplacizumab | Anti-VWF immunoglobulin fragment | N/A | New and relapsed | Single arm, caplacizumab and immunosuppression without PEX | MAYARI; Caplacizumab and Immunosuppressive Therapy Without Firstline Therapeutic Plasma Exchange in Adults With Immune-mediated Thrombotic Thrombocytopenic Purpura (2025)13,42 NCT05468320 Phase III* |
| 2 | Caplacizumab | Anti-VWF immunoglobulin fragment | 21 | New and relapsed | Single arm | A Trial of Caplacizumab in Japanese Patients With Acquired Thrombotic Thrombocytopenic Purpura (aTTP) (2023)43 NCT04074187 Phase II/III |
| 3 | Caplacizumab | Anti-VWF immunoglobulin fragment | 104 | New and relapsed | Follow-up study | Post-HERCULES; Follow-up Study for Patients Who Completed Study ALX0681-C301 (2022)44,45 NCT02878603 |
| 4 | Caplacizumab | Anti-VWF immunoglobulin fragment | 145 | New and relapsed | Randomized, double blind and placebo controlled | HERCULES; Phase III Trial With Caplacizumab in Patients With Acquired Thrombotic Thrombocytopenic Purpura (2019)46–48 NCT02553317 Phase III |
| 5 | Caplacizumab | Anti-VWF immunoglobulin fragment | 75 | New and relapsed | Randomized and placebo controlled | TITAN; Study to Assess Efficacy and Safety of Anti-von Willebrand Factor (vWF) Nanobody in Patients With Acquired Thrombotic Thrombocytopenic Purpura (aTTP) (2012)49–51 NCT01151423 Phase II |
| 6 | ADAMTS13, recombinant-krhn | rADAMTS13 | 28 | New and relapsed | Randomized, placebo controlled and double blind | SOAR-HI; Study of rADAMTS-13 (SHP655) in the Treatment of Participants With Acquired Thrombotic Thrombocytopenic Purpura (aTTP) (2023)52 NCT03922308 Phase II |
| 7 | Magnesium sulphate | Multiple mechanisms | 74 | New and relapsed | Randomized, double blind and placebo controlled | MAGMAT; Magnesium Sulfate in Thrombotic Thrombocytopenic Purpura in Intensive Care (2023)53 NCT03237819 |
| 8 | IdeS | IgG-specific endopeptidase | 2 | Remission | Single arm | IdeS in Asymptomatic Antibody-Mediated Thrombotic Thrombocytopenic Purpura (TTP) Patients (2019)54 NCT02854059 Phase II† |
| 9 | ARC1779 | VWF-binding aptamer | 7 | New and relapsed | Randomized, double blind placebo controlled | Clinical Outcome Study of ARC1779 Injection in Patients With Thrombotic Microangiopathy (2012)55 NCT00726544 Phase II‡ |
| 10 | ARC1779 | VWF-binding aptamer | 7 | New and relapsed | Dose ranging Single arm | ARC1779 Injection in Patients With Von Willebrand Factor-Related Platelet Function Disorders (2011)56 NCT00632242 Phase I/II |
| 11 | Rituximab | Anti-CD20 antibody | 12 | Remission | Single arm Pre-emptive rituximab 1,400 mg for ADAMTS13 activity <20 IU/dL | Delrue (2021)5 |
| 12 | Rituximab | Anti-CD20 antibody | 19 | New and relapsed | Single arm rituximab 100 mg subcutaneous for 5–9 days | Low Dose Rituximab in Thrombotic Thrombocytopenic Purpura (2019)57 NCT01554514 Phase II |
| 13 | Rituximab | Anti-CD20 antibody | 24 | R/R | Single arm 2–3 doses rituximab based on lymphocyte depletion | PTTritux; Rituximab in Adult Acquired Idiopathic Thrombotic Thrombocytopenic Purpura (2016)58 NCT00907751 Phase II |
| 14 | Rituximab | Anti-CD20 antibody | 6 | R/R | Single arm | Investigator Initiated Clinical Trial of Rituximab for Thrombotic Thrombocytopenic Purpura (2016)59,60 JMA-IIA00160 Phase II |
| 15 | Rituximab | Anti-CD20 antibody | 40 | R/R | Single arm | Rituximab in Patients With Relapsed or Refractory TTP-HUS (2015)61 NCT00531089 Phase II |
| 16 | Rituximab | Anti-CD20 antibody | 22 | Exacerbation or refractory | Single arm | Froissart (2012)62 |
| 17 | Rituximab | Anti-CD20 antibody | 40 | New and relapsed | Single arm | The Use of Rituximab in Acute Thrombotic Thrombocytopenic Purpura (TTP) (2011)63 NCT00937131 |
Summary of clinical trials in patients including immune TTP from 2010 to 2025.
*Completed, results not yet published.
†Terminated; cited reason: sponsor review of initial results demonstrates a non-favourable risk benefit.
‡Terminated; cited reason: enrollment into the study was slower than expected.
ADAMTS13 = a disintegrin and metalloproteinase with thrombospondin-1-like motifs, 13th member; CD20 = B-lymphocyte cell-surface molecule; IdeS = IgG-degrading enzyme of Streptococcus pyogenes; IgG = immunoglobulin G; MOA = mechanism of action; PEX = plasma exchange; rADAMTS13 = recombinant ADAMTS13; R/R = relapsed or refractory; TTP = thrombotic thrombocytopenic purpura; VWF = von Willebrand factor.
Table 3: Studies in congenital thrombotic thrombocytopenic purpura alone64–66
| # | Treatment | MOA | N | Study population | Study design | Trial name, trial identifier, year and phase |
| 1 | ADAMTS13, recombinant-krhn | rADAMTS13 | 48 | Remission | Open-label crossover comparing prophylaxis with standard therapy | A Study of BAX 930 in Children, Teenagers, and Adults Born With Thrombotic Thrombocytopenic Purpura (TTP) (2024)64 NCT03393975 Phase III |
| 2 | ADAMTS13, recombinant-krhn | rADAMTS13 | 15 | Remission | Dose escalation | Phase 1 Dose Escalation, Single Dose Study to Assess Safety and Pharmacokinetics of BAX930 in Hereditary Thrombotic Thrombocytopenic Purpura (TTP) (2017)65 NCT02216084 Phase I |
| 3 | ARC1779 | VWF-binding aptamer | 3 | Chronic/recurrent | Partial crossover design | ARC1779 Injection in Patients With Von Willebrand Factor-Related Platelet Function Disorders (2011)66 NCT00632242 Phase I/II |
Summary of clinical trials in patients with congenital TTP alone from 2010 to 2025.
ADAMTS13 = a disintegrin and metalloproteinase with thrombospondin-1-like motifs, 13th member; MOA = mechanism of action; rADAMTS13 = recombinant ADAMTS13; TTP = thrombotic thrombocytopenic purpura; VWF = von Willebrand factor.
Figure 3: Summary of progress in adjuvant therapies and FDA approvals
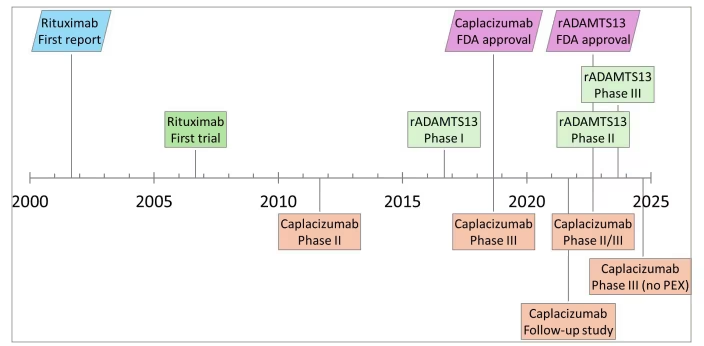
Timeline of completed trials evaluating caplacizumab and rADAMTS13, with respective FDA approvals. The initial case series of rituximab in TTP and first clinical trial are included for comparison
rADAMTS13 = a disintegrin and metalloproteinase with thrombospondin-1-like motifs, 13th member; FDA = US Food and Drug Administration; PEX = plasma exchange; TTP = thrombotic thrombocytopenic purpura.
Key therapies by category
Anti-von Willebrand factor therapies
Caplacizumab is an immunoglobulin G (IgG) fragment nanobody targeting the A1 domain of VWF, which mitigates end-organ damage by interfering with the interaction between VWF and platelet GP1b, thereby halting the formation of long chains of platelets. Caplacizumab is administered as a daily subcutaneous injection during PEX and for 30 days thereafter.
In two randomized controlled trials, HERCULES (Phase III Trial With Caplacizumab in Patients With Acquired Thrombotic Thrombocytopenic Purpura; ClinicalTrials.gov identifier: NCT02553317) and TITAN (Study to Assess Efficacy and Safety of Anti-von Willebrand Factor (vWF) Nanobody in Patients With Acquired Thrombotic Thrombocytopenic Purpura (aTTP); ClinicalTrials.gov identifier: NCT01151423), caplacizumab demonstrated a reduced time to platelet response, fewer total days of PEX and reduced recurrences when compared with placebo in acute TTP.46,49 These results are similar to a Japanese phase II/III study with a single-arm design (A Trial of Caplacizumab in Japanese Patients With Acquired Thrombotic Thrombocytopenic Purpura (aTTP); ClinicalTrials.gov identifier: NCT04074187).43 All trial designs included patients with newly diagnosed or recurrent TTP. Notably, in both studies, caplacizumab was associated with (1) a reduction in the number of exacerbations, defined as recurrences within 30 days of PEX cessation, and (2) an increase in relapses, defined as recurrences after 30 days of PEX cessation. These data suggest that caplacizumab may additionally shift TTP recurrences, changing exacerbations into relapses. A clinically significant increase in mild to moderate mucocutaneous bleeding was noted, 54% compared with 38% in placebo. Additional follow-up over 3 years demonstrated a similar safety profile.44,45
The International Society on Thrombosis and Haemostasis (ISTH) 2020 guidelines now recommend the addition of caplacizumab during acute events as soon as high clinical suspicion occurs, potentially without waiting for confirmatory ADAMTS13 assay results.7
A recent phase III trial (MAYARI; Caplacizumab and Immunosuppressive Therapy Without Firstline Therapeutic Plasma Exchange in Adults With Immune-mediated Thrombotic Thrombocytopenic Purpura; ClinicalTrials.gov identifier: NCT05468320) designed to evaluate caplacizumab and immunotherapy in iTTP without PEX has been completed as of January 2025 with results pending.13,42
B-cell depleting agents
Rituximab is an anti-CD20 chimeric monoclonal antibody, which depletes lymphocytes via complement-mediated cytotoxicity and thus interferes with the development of autoantibodies to ADAMTS13. Accordingly, it has no role in congenital TTP. Although it is not approved by the FDA for the treatment of iTTP, off-label use has been adopted following initial case reports in 2002.67 The first clinical trial evaluating rituximab in iTTP reported results in 2007 and evaluated the addition of rituximab to PEX. Of 25 patients with refractory or relapsing disease, all patients reached remission in a median of 11 days.68
Over the past 15 years, studies have evaluated the role of rituximab in acute TTP, optimal dosing strategies and prevention of recurrences.62 Two single-arm phase II trials in the UK (The Use of Rituximab in Acute Thrombotic Thrombocytopenic Purpura (TTP) ClinicalTrials.gov identifier: NCT00937131) and Canada (Rituximab in Patients With Relapsed or Refractory TTP-HUS; ClinicalTrials.gov identifier: NCT00531089) explore the addition of rituximab to standard therapy in acute iTTP. These data show that the addition of rituximab improved response rates and reduced recurrences compared with historical controls.61,63 A Japanese phase II study evaluating rituximab in the acute setting (Investigator Initiated Clinical Trial of Rituximab for Thrombotic Thrombocytopenic Purpura; Japan Registry of Clinical Trials identifier: jRCT2091220160, Japan Medical Association Center of Clinical Trials identifier: JMA-IIA00160) demonstrated improvement in laboratory parameters but did not meet the primary endpoint of response rate, presumably due to its smaller study size.59
Typical dosing schedules include rituximab 375 mg administered intravenously once weekly for four doses. Subsequent trials on rituximab in the acute phase (Low Dose Rituximab in Thrombotic Thrombocytopenic Purpura; ClinicalTrials.gov identifier: NCT01554514, PTTritux; Rituximab in Adult Acquired Idiopathic Thrombotic Thrombocytopenic Purpura; ClinicalTrials.gov identifier: NCT00907751) have assessed dosing based on lymphocyte depletion and subcutaneous administration, with similar clinical outcomes to standard dosing.57,58
Delrue et al. explored the efficacy of subcutaneous pre-emptive rituximab for patients in remission with persistent low ADAMTS13 levels <10% in remission.5 These results in 92 patients demonstrate a reduced relapse rate (15%) and improved ADAMTS13 activity compared with historical control with a 74% relapse rate after 7 years.
ISTH 2020 guidelines now recommend the addition of rituximab to steroids and PEX in acute iTTP, as well as the incorporation of rituximab for iTTP in remission for patients who develop a low ADAMTS13 activity.7
Recombinant ADAMTS13
Recombinant ADAMTS13 protein (rADAMTS13), also designated as ADAMTS13 recombinant-krhn, apadamtase alfa, BAX930, SHP655 and TAK755, replaces the depleted ADAMTS13 protein. In the acute phase, this treatment is given as a daily infusion, concomitantly with PEX until 2 days after the acute TTP event is resolved. For those receiving prophylaxis, treatment may be given once every week or once every other week.
A phase I study in patients with cTTP in remission (Phase 1 Dose Escalation, Single Dose Study to Assess Safety and Pharmacokinetics of BAX930 in Hereditary Thrombotic Thrombocytopenic Purpura (TTP); ClinicalTrials.gov identifier: NCT02216084) demonstrated that rADAMTS13 restores ADAMTS13 activity.65 A phase II randomized controlled trial SOAR-HI; Study of rADAMTS-13 (SHP655) in the Treatment of Participants With Acquired Thrombotic Thrombocytopenic Purpura (aTTP); ClinicalTrials.gov identifier: NCT03922308) demonstrated similar findings in acute iTTP. In this double-blinded study, patients were randomized to standard of care with placebo or rADAMTS13 in the acute setting, followed by 3 months of placebo or rADAMTS13 supplementation based on activity levels. Key findings included increased ADAMTS13 exposures. Development of anti-rADAMTS13 antibodies occurred; however, in remission, platelet counts improved with therapy regardless of the neutralizing antibody level.52 Additional results have not yet been published as of September 2025.
NCT03393975 (A Study of BAX 930 in Children, Teenagers, and Adults Born With Thrombotic Thrombocytopenic Purpura; ClinicalTrials.gov identifier: NCT03393975) was a phase III crossover trial comparing rADAMTS13 prophylaxis with on-demand treatment for patients with cTTP in remission.64 Forty-eight patients were randomized to standard-of-care prophylaxis with blood products, or with rADAMTS13 for 6 months followed by crossover. While one acute and five subacute TTP events occurred in patients receiving standard therapies, none occurred on rADAMTS13 therapy. No bleeding adverse events were considered related to trial treatment, and no neutralizing antibodies developed.
Following the results of the above studies, rADAMTS13 has been approved by the FDA in the prophylactic and on-demand setting for cTTP in 2023. The 2025 update to the ISTH guidelines now includes a strong recommendation to use recombinant ADAMTS13 over plasma infusion for prophylaxis during remission. In the prophylactic setting, rADAMTS13 remains attractive due to the shorter infusion time and the possibility of home infusions compared with prophylaxis with plasma infusion. While early data indicate a favourable safety profile, further research is needed to evaluate long–term outcomes and target therapeutic levels for ADAMTS13 in the prophylactic setting.
Several additional studies using rADAMTS13 remain ongoing (Table 4). A continuation study of patients enrolled in NCT03393975 (A Study of BAX 930 in Children, Teenagers, and Adults Born With Thrombotic Thrombocytopenic Purpura (TTP); ClinicalTrials.gov identifier: NCT03393975) is open-ended, designed to assess long–term safety and efficacy for patients receiving prophylactic rADAMTS13 compared with on-demand treatment.41 Further, NCT05714969 (A Study of TAK-755 (rADAMTS13) With Little to No Plasma Exchange (PEX) Treatment in Adults With Immune-mediated Thrombotic Thrombocytopenic Purpura (iTTP); ClinicalTrials.gov identifier: NCT05714969) is a randomized study in patients with acute iTTP with a double-blind design. rADAMTS13 will be compared with minimal to no PEX in the treatment of iTTP.14 These extension trials are critical to evaluate long-term outcomes, durability of clinical response and the potential for the development of neutralizing antibodies. Results may inform the role of rADAMTS13 in long-term prophylaxis and acute treatment strategies.
Other therapies
Magnesium sulphate was evaluated in TTP, given in vitro evidence that it may potentiate the effect of ADAMTS13 and inhibit the interaction between VWF and platelets. A randomized, double-blind study (MAGMAT; Magnesium Sulfate in Thrombotic Thrombocytopenic Purpura in Intensive Care; ClinicalTrials.gov identifier: NCT03237819) in 74 patients with a clinical diagnosis of TTP did not demonstrate superiority in the time to platelet normalization, response rate or adverse reactions.53
ARC1779 is an aptamer oligonucleotide that binds the A1 domain of VWF, interfering with its ability to interact with platelets. Thus, like caplacizumab, its activity may be measured through the inhibition of VWF. ARC1779 was given as a continuous infusion until the remission of TTP in the adjuvant setting.
In phase I/II studies (ARC1779 Injection in Patients With Von Willebrand Factor-Related Platelet Function Disorders; ClinicalTrials.gov identifier: NCT00632242) of patients with immune and congenital TTP, ARC1779 reduced VWF activity measured by enzyme-linked immunosorbent assay.56,66 Some patients demonstrated improved platelet counts. PTT increases without increases in bleeding were identified.56 A subsequent phase II randomized controlled trial (Clinical Outcome Study of ARC1779 Injection in Patients With Thrombotic Microangiopathy; ClinicalTrials.gov identifier: NCT00726544) was terminated due to slow accrual, and further development of the drug was discontinued. In this study, seven patients received the study medication and two received a placebo without any safety events.55
The IgG-degrading enzyme of Streptococcus pyogenes (IdeS) is a streptococcal endopeptidase which cleaves IgG molecules and has been explored in diseases mediated by IgG-driven human disease. A phase II single-arm trial in patients with TTP in remission (A Phase II Pilot Study to Evaluate the Safety, Tolerability, Efficacy, Pharmacodynamics and Pharmacokinetics of IdeS in Asymptomatic Antibody-Mediated Thrombotic Thrombocytopenic Purpura (TTP) Patients With Low ADAMTS13 Activity; ClinicalTrials.gov identifier: NCT02854059) was terminated due to toxicity; both patients developed acute serum sickness on days 6 and 7.54 Ultimately, this medication has continued in clinical development in other indications. Now recognized as imlifidase (Idefirix, Hansa Biopharma, Lund, Sweden), this medication has been approved in the EU for the desensitization of highly sensitized patients with a positive crossmatch against an available deceased donor kidney transplant.69
Discussion
The addition of caplacizumab and rituximab to PEX and corticosteroids for acute TTP has changed practice and improved outcomes. Ongoing studies may inform optimal therapies in the treatment of acute TTP and further define prophylactic treatment for patients in remission. rADAMTS13 demonstrates promise in the management of acute TTP and in prophylaxis; however, ongoing studies are needed to define its role in the current treatment landscape. Data regarding safety, sustained activity and the development of neutralizing antibodies remain under active investigation. Should the following trials confirm consistent long–term benefit, rADAMTS13 may serve as a powerful adjunct therapy in iTTP and challenge the role of PEX in cTTP.
Other therapies have been explored without success in clinical trials. Magnesium sulphate has not been shown to improve outcomes. Development of IdeS in this setting was stopped due to toxicity. Further, the development of ARC1779 was halted due to slow trial accrual.
Drug development in TTP faces several barriers. Owing to its low incidence and high mortality, often trials are run as single-arm studies and may face difficulty with accrual. Sufficiently powered studies may require multinational or registry-based designs. Optimal endpoint selection may be challenging, as markers like time to platelet normalization and ADAMTS13 activity may not predict long-term clinical benefit. Ethical constraints often restrict trial design options and the use of placebo in acute settings. For patients who may be intolerant or present with a contraindication to a therapy used in TTP, few options are available. Case reports describe the use of alternative agents for B cell lymphodepletion, such as ofatumumab, bortezomib and vincristine, but these are difficult to explore in a clinical trial setting. Establishing equipoise for studies involving these agents may be challenging, especially when standard therapies are well established and carry less risk.
Conclusions
Multiple agents have been explored in the clinical trial setting for immune and congenital TTP. The rare disease incidence is a barrier to randomized trial design. Adjuvant therapies to improve remission durability and relapse rate, as well as those for prophylaxis and long-term management are active areas of development.