Polycythaemia vera (PV) is a chronic myeloproliferative neoplasm characterised by unregulated proliferation of red blood cells, and sometimes leukocytes and platelets in the absence of a recognisable physiological stimulus.1–4 Increased red blood cell mass leads to hyperviscosity of the blood and increased risk of thrombosis. In addition, patients may have a high symptom burden that includes intractable pruritus, fatigue and altered quality of life (QoL).5,6 If inadequately controlled, the condition can lead to serious complications including increased risk of cardiovascular events, together with the development of splenomegaly as the disease progresses.1 The disease progresses slowly and is associated with a reduced life expectancy;3 the median survival of PV patients is 14.1 years,7 and is lower in patients who are resistant or intolerant to hydroxyurea (HU), as well as patients at high risk of thrombosis. In addition, PV may transform into acute myelogenous leukaemia (AML) in up to 15% of patients every 10 years7,9 or myelofibrosis (MF) in up to 10% of patients every 10 years.10–12
PV is an uncommon but not so rare condition: its annual incidence is approximately 2.8 per 100,000 population of men and approximately 1.3 per 100,000 population of women. The prevalence of PV is approximately 22 cases per 100,000 people.3 The median age at presentation is in the sixth decade but one third of patients with PV are under 50 years of age.13
Despite advances in the treatment of PV, the symptom burden is often underestimated by physicians and is poorly managed, resulting in impaired QoL,14 restricted participation in social events and physical activity.6 Most therapies in PV appropriately aim to reduce thrombotic risk, which is the first cause of mortality and morbidity in the short term, but do not address PV symptoms. Recently, the Janus kinase (JAK) inhibitor ruxolitinib was approved for patients with PV who are intolerant to or resistant of HU, the most widely used first-line therapy for high-risk patients with PV. However, ruxolitinib is not commonly used in clinical practice15 and there is a need to identify patients who will derive the most benefit from this therapy and to define inadequately controlled PV. This article aims to examine what constitutes inadequate control in PV and explores current treatment strategies.
Current strategies for the management of polycythaemia vera
The primary aim of present PV therapy is not to eliminate disease, but to reduce the incidence of thrombotic events while minimising the risk of haematological transformation to AML or MF.16 Patients with PV are therefore treated according to their risk for thrombosis: patients aged <60 years and with no history of thrombosis are considered low risk while those age >60 years and/ or with history of thrombosis are high risk (see Table 1).17–19 Some centres will also treat according
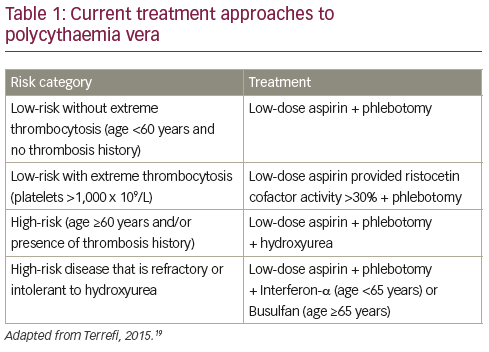
to symptoms when they have an impact on patient’s life, i.e. itching, splenomegaly, fatigue etc. Low-risk patients are currently managed by phlebotomy and low-dose aspirin.19 Phlebotomy has been associated with increased survival in PV though with an increased thrombotic risk during the first three years of treatment.20 However, phlebotomy has little impact on PV symptoms,21 and has recently been associated with an elevated risk of thrombosis.22 Among possible symptomatic treatments, paroxetine has been associated with relief of PV-associated pruritus, one of the most common symptoms of PV.23
For high-risk patients, the currently proposed management includes cytoreductive therapy, most commonly using HU.16 The multicentre Cytoreductive therapy in Polycythemia Vera (CYTO-PV) trial established the threshold of 45% of haematocrit as the target of therapy in PV, achieved either with phlebotomy or cytoreductive drug. This study randomised 365 patients with PV to high-intensity treatment aimed at maintaining a low target haematocrit of <45% or to low intensity treatment with a higher target haematocrit of 45–50%. At a median follow-up of 31 weeks, the primary endpoint (death from cardiovascular causes or major thrombotic events) was reported in 2.7% in the low haematocrit group and 9.8% of the high haematocrit group (hazard ratio in the high haematocrit group, 3.91; 95% confidence interval [CI], 1.45 to 10.53; p=0.007).24 In addition, an analysis of data from the CYTO-PV study and the Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF) study5 found that symptom burden was more severe in patients with haematocrits >45%. However, strict maintenance of a haematocrit below 45% was also associated with loss of concentration, insomnia, weight loss, night sweats, pruritus and significantly higher levels of fatigue, most likely a result of iron deficiency.25
In addition to controversy regarding the treatment goals of cytoreductive therapy, concerns persist around the use of HU. Although recent retrospective studies or registry data have not found a link between HU use and leukaemic transformation,7,26 some study findings suggest that long-term HU treatment increases the risk of transformation to AML, leading to unease among both patients and haematologists.12,27–32 In addition, around 20 to 25% of patients have an inadequate response to HU or have unacceptable side effects at the doses required for haematocrit control.33,34 Dose titration of HU up to 2 g per day to achieve complete response is acknowledged under current recommendations16 and can improve the therapeutic response, but few patients can tolerate this dosage in clinical practice. Toxicities associated with HU include leukopenia, thrombocytopenia, painful leg ulcers, skin cancers, extensive dermatitis and fever.35
Resistance to HU was shown to be associated with disease transformation and reduced survival: compared with other patients with PV, patients with resistance to HU have a 5.6-fold increase in the risk of death.34 However, therapeutic options for PV patients with resistance or intolerance to HU are limited, given that no drugs had approval in this particular setting, except pipobroman in few countries (France and Italy). As a result, many patients continue to receive HU despite an inadequate response or significant adverse effects. Clinical trials on the optimum treatment to improve survival of PV are difficult to perform because of the prolonged survival of patients with PV (typically one to two decades), necessitating an extended follow-up period in order to fully assess the benefits and risks associated with a therapeutic intervention. In addition, the rarity of the condition limits access to sufficient numbers of patients at individual treatment centres.28
The use of interferon alpha (IFN-α) in the treatment of PV was first proposed in 1993.36 INF-α inhibits the growth of the abnormal clone in patients with myeloproliferative disorders, has a direct inhibiting effect on bone marrow fibroblast progenitor cells, and antagonises the action of platelet-derived growth factor, transforming growth factor-β, and other cytokines that may be involved in the development of MF.32,37 It can achieve molecular responses and is recommended by European LeukemiaNet (ELN) as possible first-line therapy for PV.16
Despite its proven clinical efficacy, the use of IFN-α has been limited by toxicity, and prolonged evaluation of IFN-α in PV is difficult due to high discontinuation rates.29,30 Lower doses of IFN have reduced the toxicity while maintaining efficacy.38 Reduced administration frequency has also been associated with improved tolerability.39 In addition, sustainedrelease preparations such as pegylated IFN-α 2a (Pegasys®, Genentech, California, US) and ropeg IFN-α 2b have been associated with excellent clinical activity and high rates of molecular response, in patients with PV.40–44 However, while pegylated forms are better tolerated than standard IFN-α, toxicity remains a problem: in an analysis of the longterm (median 42 months) use of peg-IFN-α in 103 patients with highrisk essential thrombocythemia, discontinuation was reported in 47% of patients. Of these, 59% discontinued due to toxicity.45 In addition, IFN is less convenient to administer (by subcutaneous injection) than an orally available drug. The use of peg-IFN-α allows a weekly or twicemonthly (with ropeg IFN-α 2b) injection regimen, which is preferred by patients and can also be adjusted for a flexible dosing schedule, for example, when a patient is on holiday. In addition, since peg-IFN-α is not a cytotoxic therapy, patients typically have a more positive emotional response to treatment.
Although the ELN guidelines recommend using either HU or IFN (off label) as first-line therapy of high-risk PV,16 other experts proposed some limitations in the use of this drug. For example, Passamonti recommends peg-IFN-α in high-risk females with childbearing potential (but avoiding peg-IFN-α while pregnant); high-risk young patients who refuse HU because of the fear of evolution to leukaemia; and as second-line therapy after HU intolerance or resistance in high-risk young people.46 However, others consider peg-IFN-α safe in pregnancy, highlighting the need for larger clinical studies of peg-IFN-α in PV. To date, efficacy and safety data on IFN is restricted to small cohort studies. Two randomised controlled studies are ongoing47,48 but no data are currently available. The treatment is not approved for PV and is not available for this indication in many European countries.
The only possible curative treatment for PV is allogeneic stem cell transplantation, and this can be effective in patients whose disease has
undergone transformation to AML or MF. However, it does not represent a viable treatment option for the vast majority of patients in the chronic phase of the disease, due to the risk of complication.49–51 In addition, most patients with PV are of advanced age and have comorbidities, further increasing the risk of transplant-related mortality.
The ELN recommends IFN-α as second-line therapy of PV in patients intolerant or resistant to HU. Conversely, HU is recommended as the second-line therapy for patients who are intolerant or refractory to firstline therapy with IFN-α.16. The efficacy of IFN as second-line therapy in patients with PV who had an inadequate response to HU is unknown. In a recent clinical study, a subset of 13 HU intolerant or resistant patients received IFN: three achieved a complete haematologic response, four had haematocrit control, and none had a reduction of 35% or more in spleen size.52,53
Pipobroman54 and radioactive phosphorus (32P)55 are effective secondline options for patients with a short life expectancy, but they have been associated with a significant rate of transformation to acute leukaemia or myelodysplastic syndrome, so their use should be avoided in younger patients.7,12,26 Busulfan (Myleran®, GlaxoSmithKline, Brentford, UK) can be very effective in elderly patients,56 especially if the patient has concerns regarding medication; a maintenance dose (5–10-day course of 4 mg/day) may be administered weekly. The drug is well tolerated,57 and can also be given as a pulse dose for 5–10 days when the platelet count rises over desired range. In some cases it may only require an average of four pulses a year. While some studies suggest that bulsufan is associated with a significant rate of leukaemic transformations,26,56 a large (n=1,545) study found no such association.7 However, busulfan is clearly mutagenic and carcinogenic, as reported in the summary of product characteristics of this drug.
Other novel therapies, including histone deacetylase inhibitors58–60 and heat-shock protein-90 inhibitors,61 are currently being investigated in PV.
Ruxolitinib, a new second-line therapy for the treatment of polycythaemia vera
Ruxolitinib (Jakavi®, Novartis Oncology, known as Jakafi® and marketed by Incyte Corporation in the US) is an inhibitor of JAK1 and JAK2, which are non-receptor tyrosine kinases, approved for several years for the treatment of MF.62 Approximately 96% of PV patients have a mutation of
the JAK2 gene (JAKV617F),63,64 which is involved in intracellular signalling of cytokine receptors, including the receptor of erythropoietin. This mutation causes constitutive activation of the kinase and hyperactivity of the JAK-STAT pathway, resulting in uncontrolled growth of blood cells. Ruxolitinib is administered orally at a starting dose of 10 mg twice daily in PV.65
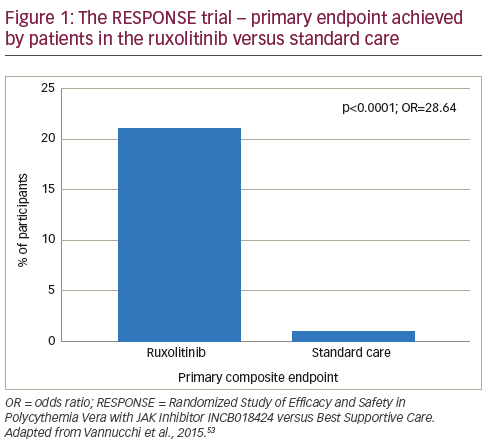
In a phase II study, ruxolitinib was associated with rapid and sustained improvements in PV symptoms.65 Following these findings, the Randomized Study of Efficacy and Safety in Polycythemia Vera with JAK Inhibitor INCB018424 versus Best Supportive Care (RESPONSE) trial was initiated.52 This phase III study randomised patients (n=222, with splenomegaly and needed phlebotomy for adequate haematocrit control) to ruxolitinib (n=110) or best available therapy (n=112). At week 32, patients were given the option to cross over to ruxolitinib if the primary end point was not met or could switch later in the case of disease progression. Among patients assigned to standard care, 96 (85.7%) crossed over to ruxolitinib at or after week 32. At week 32, the proportion of patients who achieved the primary composite end point (haematocrit control and a reduction of 35% or more in spleen volume from baseline) was significantly higher among patients in the ruxolitinib group than in the standard-therapy group (20.9% versus 0.9%, p<0.001; see Figure 1), with 77% of patients meeting at least one component of the composite endpoint. A complete haematologic remission occurred in 24% of patients in the ruxolitinib group versus 9% of those in the standard-therapy group (p=0.003). In addition, a reduction of at least 50% in the in the MPN-SAF at week 32 was reported in 49% of the ruxolitinib group versus 5% of those receiving standard care.52
Ruxolitinib was generally well tolerated: grade 3 or 4 anaemia occurred in 2% of patients taking ruxolitinib versus 0% receiving standard care, grade 3 or 4 thrombocytopenia occurred in 5% versus 4%, and grade 1–3 Herpes zoster infection in 6% versus 0%. Four patients in the ruxolitinib group and two patients in the standard-therapy group had newly diagnosed nonmelanoma skin cancer (basal-cell or squamous-cell carcinoma); all but one patient (in the standard-therapy group) had a history of non-melanoma skin cancer or precancerous skin lesions. Only one thrombotic event was observed in patients receiving ruxolitinib versus six events in the standard care group. Although not powered to determine the significance of this endpoint, this was an interesting finding that should be explored in further studies. At the study end, MF developed in three patients receiving ruxolitinib, and one patient had received a diagnosis of AML. In the standard
therapy group, MF developed in one patient assigned to standard therapy. These rates of transformation were consistent with those expected in a high-risk population of patients with PV.52
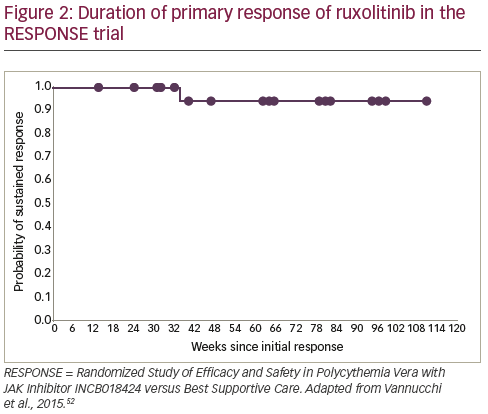
As a result of these data, ruxolitinib received approval from the European Medicines Agency66 and the US Food and Drug Administration67 for PV patients who have an inadequate response to, or cannot tolerate HU. Follow-up data indicate that ruxolitinib responses are durable and the treatment is well tolerated, with 83% of patients still receiving treatment at a median 111 weeks.6869 In addition, the ongoing Randomized Switch Study from Hydroxyurea to Ruxolitinib for RELIEF of Polycythemia Vera Symptoms (RELIEF) study is investigating the effect of a switch from HU to ruxolitinib for the relief of PV symptoms.70
Several other JAK inhibitors have been evaluated in clinical trials for PV, including momelotinib,71 pacritinib72 and fedratinib.73 However, the development of fedratinib has been discontinued, and the programme of pacritinib is currently on hold, waiting for more detailed safety results of phase III studies.
Definition of patients with inadequately controlled polycythaemia vera
A recent prospective study (n=1,334), including a mixture of patients with current and prior therapy use, found the presence of prior HU use, the need for phlebotomy or palpable splenomegaly was associated with an unacceptably high symptom burden, regardless of PV risk category. Patients with all three features had the highest symptom burden.74 The authors suggested that patients with PV who had any of these three features might benefit from ruxolitinib treatment.
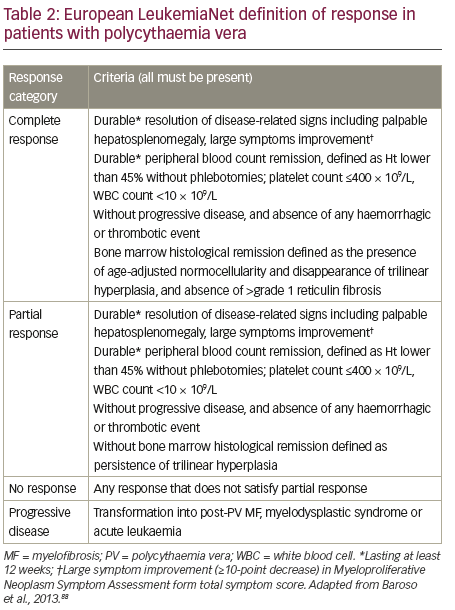
Response to PV therapy is currently defined according to ELN criteria (see Table 2). We propose that the definition of inadequate response to first-line therapies could be further refined to incorporate five patient types who do not have an adequate response to HU and may benefit from ruxolitinib treatment.
Patients with a high symptom burden after taking hydroxyurea
Symptom burden can remain high despite treatment with HU. In a 2007 internet survey of 405 patients with PV who were receiving standard treatments, symptoms included: fatigue (85%), pruritus (65%), night sweats (49%), bone pain (43%), fevers (13%), undesired weight loss (10%) and spleen pain (4%). All of these had a significant negative impact on patients’ overall QoL and ability to work.6 A study of patients with myeloproliferative neoplasms (n=1,470) found that distinct clustering of symptom burden exists within PV with significant heterogeneity of symptoms and with significant symptom burdens among all risk categories. The MPN-SAF total symptom score (MPNSAF TSS; MPN-10) is a well-validated instrument for the evaluation of the symptom burden that should be used to better identify patients in need of therapy.
Patients who require frequent phlebotomy
In the RESPONSE study, phlebotomy dependence was defined as two or more phlebotomies within 24 weeks prior to screening and at least one within 16 weeks before screening.52 While therapeutic phlebotomy has become the mainstay of treatment, it has limitations. Patients may be intolerant or have a low acceptance, and it may be difficult to gain peripheral vein access.75 In addition, phlebotomy can cause physical and psychological distress if venous access is difficult, and not all patients can tolerate venesections – many feel quite unwell afterwards or require intravenous fluid replacement.
Phlebotomy has been associated with complications, including reactive thrombocytosis during the first three months of treatment.20 Venesections may also be inconvenient in terms of taking time out of work, the need for hospital visits as well as the potential for bruising and scarring from frequent phlebotomy. Furthermore, the need for repeated phlebotomy can lead to iron deficiency, symptoms of which include restless leg syndrome76 and anxiety,77 and can further induce an increase in platelet counts. Repeated phlebotomies under HU therapy have also been associated with an increased risk of thrombosis.22
Patients with elevated leukocyte count
Leukocyte activation has been associated with activation of endothelial cells and pro-coagulant responses at sites of vascular injury,78 as well as being associated with thrombosis and major haemorrhage in other myeloproliferative neoplasms.79 In addition, leucocytosis is associated with decreased overall survival in PV.7 In the CYTO-PV study, the platelet count was not significantly different between treatment arms, but the leukocyte (WBC) count was significantly higher in the high-haematocrit group.24 These data highlight the importance of leukocyte count in identifying inadequate response in PV. A sub-analysis of the CYTO-PV study showed that the risk of thrombosis was increased at WBC count >7 x 109/L, which became statistically significant when the WBC count exceeded 11 x 109/L, and suggested that this should be taken into account when evaluating response to therapy in PV.80 Elevated platelet counts may also be an indicator of inadequately controlled PV.
Intolerance to hydroxyurea or interferon
Clinical data suggest that up to 20% of patients develop toxicities to HU, including painful leg ulcers, skin cancers, extensive dermatitis, hyperpigmentation, alopecia, fever and significant fatigue,35,81,82 which may necessitate dose reduction or drug discontinuation. A recent study of 890 patients with PV identified resistance/intolerance to HC was recorded in 15.4% of patients. This consisted of: need for phlebotomies (3.3%), uncontrolled myeloproliferation (1.6%), failure to reduce massive splenomegaly (0.8%), development of cytopenia at the lowest dose of HC to achieve a response (1.7%) and extra-haematological toxicity (9%). Cytopenia was also associated with a risk of transformation to MF and acute leukaemia.83 IFN treatment, currently mostly using pegylated forms, has well-defined contra-indications and is associated with toxicities including exacerbation of autoimmune disorders, depression, neuropathy, hyperthyroidism, heart and ocular disease, leading to permanent discontinuation in about 25% of patients.40–44,84
Symptomatic splenomegaly
Another burdensome symptom is symptomatic splenomegaly. Palpable splenomegaly has been reported in up to 40% of patients with PV6,7 and can lead to severe splenomegaly, with symptoms including early satiety, bloating, pain, portal hypertension, weight loss and exacerbation of cytopenias.6 Severe splenomegaly at diagnosis is also associated with decreased survival in PV.85 Real-world data on the use of ruxolitinib in patients with lower-risk MF suggest that ruxolitinib is well tolerated and is associated with marked reduction in splenomegaly.86
Summary and concluding remarks
Despite advances in treatment, the symptom burden of patients with PV remains high and is often underestimated by physicians focusing on vascular events. The traditional stratification of patients as ‘highrisk’ and ‘low-risk’ has limitations, and in order to optimise outcomes, it is necessary to redefine inadequate response to treatment. Certain groups of patients with PV may be considered inadequate responders to HU. These include high-risk patients who retain a high symptom burden after HU, require frequent phlebotomy, have an elevated leukocyte count, are intolerant to HU or IFN-α, or have symptomatic splenomegaly. For patients with these factors, ruxolitinib may be a beneficial secondline treatment option, and such expanded indications for the use of ruxolitinib could reduce the symptom burden and improve the QoL of many PV patents.
The most important safety concern of younger patients with PV is the potential for transformation into AML or MF. Evidence to date suggests that ruxolitinib is not associated with these risks, but there is a need for further data, including larger clinical trials and real-world data, to assess the long-term risk. The availability of ruxolitinib has raised the hypothesis that disease elimination may be possible in future by combining a JAK inhibitor and a low dose of IFN; a recent case report shows a rapid decline in the JAK2V617F allele burden following combination therapy, an impressive finding that warrants further investigation.87 In conclusion, the development of ruxolitinib represents a major breakthrough for patients with PV as it has the potential to reduce disease burden and substantially improve QoL in many patients with inadequately controlled PV for whom effective treatment opportunities have been limited.