
Current treatment landscape in Canada
Waldenström’s macroglobulinaemia (WM) is a rare haematological disorder with an incidence of about three cases per million, leading to approximately 150 new cases per year in Canada.1,2 WM is characterized by a clonal lymphoplasmacytic infiltration of the bone marrow and the presence of an IgM monoclonal protein.3,4 Symptomatic disease can arise from direct tumour infiltration of tissues such as nodes and spleen, or from IgM-related autoimmune activity (e.g. cytopenias, neuropathy). WM can manifest with heterogeneous clinical complications, including central nervous system involvement (Bing–Neel syndrome), peripheral neuropathy, concurrent amyloidosis and hyperviscosity. Treatment is only instituted when such complications or symptoms arise. Chemoimmunotherapy has historically been the standard of care in the treatment of WM, with combinations of rituximab plus nucleoside analogues (e.g. fludarabine, cladribine), alkylating agents (e.g. cyclophosphamide or bendamustine), or proteasome inhibitors (e.g. bortezomib) demonstrating significant antitumour activity.5 Specific combinations such as fludarabine/rituximab, cyclophosphamide/dexamethasone/rituximab, and bortezomib/dexamethasone/rituximab have gained popularity and are used variably, with reported response rates of 83–95% and median progression-free survival (PFS) of 35–51 months.6–10
Bendamustine and rituximab (BR) is the treatment of choice for frontline therapy in many countries, including Canada, based on the original Stil trial reported by Rummel et al., which compared rituximab-cyclophosphamide/doxorubicin hydrochloride/vincristine/prednisolone (R-CHOP) with BR in indolent lymphomas.11 Within this trial, a subset of 41 patients with WM achieved similar responses to either treatment, but BR led to a superior PFS (70 versus 28 months). BR was well tolerated with reduced rates of alopecia, stomatitis, paraesthesia and infections. The French Innovative Leukemia Organization (FILO) administered BR to 69 untreated WM patients and similarly reported a major response rate (MRR) of 96%, a very good partial response (VGPR) rate of 37% and a complete response (CR) rate of 19%.12 More recently, the Stil NHL7-2008 MAINTAIN trial provided further data on long-term BR outcomes.13 This was a phase III trial in which 293 patients with WM were treated with first-line BR, with responders then randomized to either maintenance rituximab or placebo. No statistically significant differences in PFS between the two arms were noted. However, whether maintenance was used or not, the reported PFS rates were impressive (83–103 months), thus emphasizing how effective the BR regimen alone is in frontline treatment of WM. Hence, most healthcare providers in Canada do not routinely use maintenance rituximab after initial BR. This practice also takes into consideration the toxicities and excess fatal events during rituximab maintenance reported in the GALLIUM trial in follicular lymphoma.14,15 With its high response rates and impressive PFS, BR remains an effective and appealing treatment choice for frontline WM therapy and is particularly attractive due to its finite treatment course.
A 6-month course of BR is, however, not without challenges. Patients with WM are elderly and can be frail, thus completing a full course of therapy can be difficult, as shown in the FILO trial where only 56% of patients completed six courses at the planned dose.12 In another trial by Dogliotti et al. in frail and elderly patients with lymphoid malignancies, only 49% of the patients received the full regimen at the standard dose of 90 mg/m2.16 In Canada, it is common practice in elderly patients to empirically reduce the daily bendamustine dose from 90 to 70 mg/m2 or to truncate the total number of BR cycles from six to four, in an attempt to improve tolerability. In support of this approach is a single institution report by Paludo et al. of 60 patients with treatment-naïve (TN) or relapsed/refractory (R/R) WM treated with BR; 52% received the full six cycles and 48% received a truncated course (23% four cycles).17 The MRR with six cycles (93%) was similar to that with four cycles (90%; p=1.0), though there was a trend towards deeper responses (CR/VGPR) with six cycles (46%) compared with four cycles (20%; p=0.25). Median PFS was not reached for either group. Furthermore, exploratory analyses from a retrospective study published by Castillo et al. showed that there was no difference in major response, deep response (VGPR or better), PFS or overall survival (OS) based on the dose of bendamustine received, whether it was 90 mg/m2 per dose or 60–70 mg/m2 per dose. Once again, no difference in the same outcomes was seen between patients receiving six cycles of bendamustine and those receiving fewer than six cycles.18
The hospital-based parenteral administration of BR may also be limiting, particularly in a large country such as Canada where many patients live in remote locales distant from cancer centres. The incurability of WM means that patients are confronted with geographical restrictions repeatedly throughout their lifetime, a challenge that is exacerbated by reduced mobility as they age. Therefore, novel oral regimens that can be administered conveniently at home are particularly important in this disease.
Finally, it should be noted that bendamustine, as an agent with both alkylator and purine analogue characteristics, may increase the risk of secondary malignancies and myelodysplastic syndrome.19,20 However, reported rates of myelodysplastic syndrome from real-world data in 1,022 patients with chronic lymphocytic leukemia (CLL) who were treated with bendamustine are reassuringly low, at 0.48% after a median of 1.7 years, though a higher incidence may be anticipated with longer follow-up.21
Molecular rationale for targeting Bruton’s tyrosine kinase in Waldenström’s macroglobulinaemia
As per the consensus treatment recommendations from the Tenth International Workshop for Waldenström’s Macrogobulinaemia (IWWM-10), chemotherapy along with rituximab, or a Bruton’s tyrosine kinase (BTK) inhibitor with or without rituximab are both listed as preferred first-line therapy options for symptomatic patients with WM.22 Ibrutinib is the first BTK inhibitor available commercially and approved in many countries, including Canada. It is currently used for the treatment of untreated or treated WM, with both molecular and clinical rationale supporting its use in this disease.
In 2012, Treon et al. first reported that over 90% of patients with WM carried a single nucleotide switch from leucine to proline in the myeloid differentiation primary response 88 (MYD88) gene at amino acid position 265 (L265P).23 MYD88 is an adaptor protein that mediates Toll-like and interleukin-1 receptors, activating interleukin-1 receptor associated kinase (IRAK1) and IRAK4 kinases, ultimately leading to nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation. The MYD88L265P mutation alters the structure of the Toll-like receptor protein, resulting in constitutive formation of the MYD88/IRAK complex leading to persistent NF-κB activation, stimulation of proliferation and suppression of apoptosis.24 When harbouring the L265P mutation, MYD88 can also bind and activate BTK, causing further activation of the Janus kinase-signalling transducer and activator of transcription 3 (JAK-STAT3) pathway via IRAK1/4 and ultimately NF-κB.25,26 This provides the molecular rationale for targeting BTK in WM.27 Although the L265P mutation accounts for most MYD88 mutations observed in WM, it is important to note that lower-frequency mutations, including V217F, S219C, M232T, S243T and S243N, may not be routinely tested at most commercial laboratories and therefore true MYD88WT may be missed.28,29
Differences in mutational status, including sensitivity and mechanisms of action to BTK inhibitors, can differ. As patients with MYD88WT will often have mutations downstream of BTK in the NF-κB pathway, they will not respond in the same way to inhibition of BTK as those with MYD88MUT.30 Mutations of CXCR4, a receptor specific to stromal-derived factor-1 (SDF-1), which is strongly chemotactic to lymphocytes, is another recurrently mutated gene in WM (30% incidence),31 occurring almost exclusively in patients with MYD88MUT disease. Various mutations in the C-terminal domain of CXCR4 have been described and patients can harbour multiple different mutations as a result of genomic instability. These mutations result in loss of regulatory serines and constitutive activation of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signalling pathway through enhanced SDF-1-triggered activation. Patients harbouring a CXCR4 mutation will respond more slowly to BTK inhibition as a result of PI3K/AKT signalling.32 As with MYD88 variants beyond the L265B mutation, CXCR4 testing is not broadly available in many countries, including Canada.
Clinical data supporting the use of Bruton’s tyrosine kinase inhibitors in Waldenström’s macroglobulinaemia
Ibrutinib
Ibrutinib is an orally administered BTK inhibitor that forms a covalent and irreversible bond with the cysteine residue at the active binding site of BTK (C481). In a pivotal phase II non-randomized trial of ibrutinib monotherapy in the treatment of 63 patients with RR WM, a 90% overall response rate (ORR), including minor responses (at least a 25% reduction of IgM paraprotein), with a rapid onset of 4 weeks was reported.33 This trial led ibrutinib to become the first drug to be approved specifically for WM in Canada. In a recent update of this trial, the 5-year PFS was 54%.34 In another phase II trial of ibrutinib monotherapy in 31 patients with TN WM, the MRR was 83%, with a VGPR rate of 20%.35 Again, responses were relatively rapid in onset, with median time to major response of 1.9 months. A recent update of this trial reported a 4-year PFS of 76%.36 The iNNOVATE trial, a phase III randomized trial of 150 TN or R/R patients, compared ibrutinib/rituximab (IR) with placebo/rituximab (R), using rituximab 375 mg/m2 given intravenously for eight weekly doses on weeks 1–4 and weeks 17–20 in both treatment groups.37 Although single-agent rituximab was generally regarded as a poor control regimen, it was at that time a commonly used real-world regimen for WM in countries such as the USA. In this study, the IR arm led to an MRR of 72% compared with 32% in the R arm, with 27% reaching a VGPR or better compared with 3%, respectively.38 After a follow-up of 54 months, the PFS favoured the IR arm (68% versus 25%).39 The utility of rituximab added to ibrutinib monotherapy remains unclear in WM, though one might generalize from the CLL literature that IR offers no survival benefit over ibrutinib monotherapy.40 These studies together confirm the impressive efficacy of ibrutinib monotherapy in WM, with virtually all patients responding and the majority remaining free of progression for over 5 years.
The pharmacokinetic profile of ibrutinib makes it particularly useful in the treatment of challenging subgroups of WM, including those with central nervous system involvement or Bing–Neel syndrome and symptomatic hyperviscosity syndrome. Ibrutinib is able to cross the blood–brain barrier41,42 and can lead to resolution of neurological symptoms and radiological findings in over 80% of patients with Bing–Neel syndrome, and can clear malignant cells from cerebrospinal fluid in 47%.43–45 Ibrutinib does not cause IgM flare, an acute but transient increase in IgM associated with rituximab and other chemotherapies, predisposing to hyperviscosity complications.33,35 In the iNNOVATE trial, transient increases in IgM levels occurred in only 8% of the patients treated with IR compared with 47% treated with rituximab alone.37 Hence, ibrutinib is considered safe to use in patients at high risk or with existing hyperviscosity.
Not all patients will do well with ibrutinib however. As previously noted, both CXCR4 and MYD88 mutational status can significantly influence response to ibrutinib. In the pivotal phase II trial of ibrutinib monotherapy in R/R WM, patients with CXCR4MUT versus CXCR4WT exhibited lower major responses (68.2% versus 97.2%) and VGPR rates (9.1% versus 47.2%), and required a longer time to major response (4.7 versus 1.8 months), respectively.46 In a recent update, there was also a trend towards a lower 4-year PFS rate in patients with CXCR4 mutations (59% versus 92%).36 These findings were further confirmed in the phase III iNNOVATE trial where patients with CXCR4MUT required longer time to major response (2.8 months) compared with patients with CXCR4WT (1.9 months), though 30-month PFS was similar in both groups.37 In a substudy of this trial where 31 patients with rituximab-refractory disease received open-label IR, the overall responses were similar for CXCR4WT and CXCR4MUT patients, although the magnitude of IgM reduction was greater and response was earlier in those with CXCR4WT.38,47 Although these data highlight the differential sensitivity of disease associated with the presence or absence of the CXCR4 mutation in patients receiving ibrutinib, it should be emphasized that patients with CXCR4MUT can still achieve prolonged disease control with ibrutinib, though one should anticipate a slower response and avoid premature discontinuation. For those rare patients with the MYD88WT genotype, significantly inferior outcomes with ibrutinib are observed. Among the four MYD88WT patients in the phase II trial in the R/R setting, none were able to achieve a major response and the median PFS was dismal at 0.4 years, far inferior to the 5-year PFS of 70% reported in patients with the CXCR4WTMYD88MUT genotype.34,46 Though numbers are far too small to make definite conclusions, these findings emphasize the importance of testing for MYD88 variants to identify this rare and potentially poor-risk subgroup with MYD88WT.
Although ibrutinib is a highly active agent for WM, as a single agent it does have its limitations. Durable responses are achieved only with continuous suppressive therapy, as deep levels of response are not achievable. In the long-term 5-year follow-up of the iNNOVATE trial, only 1% of patients achieved a complete response.39 Ibrutinib may therefore not be the treatment of choice in patients with concurrent amyloidosis or IgM-related peripheral neuropathy, where elimination of the offending monoclonal protein is a prerequisite to clinical improvement. Furthermore, although ibrutinib is generally considered to be a well-tolerated drug, about 10% of patients with WM will still discontinue therapy due to adverse events and up to 25% of patients will require dose reduction.34,48 Common toxicities (all grades), as reported in the iNNOVATE trial occurring over 2 years of treatment, included bleeding (51%), diarrhoea (28%), arthralgia (24%), nausea (21%), asthenia (16%), hypertension (13%), atrial fibrillation (12%) and pneumonia (9%).37 In the study, atrial fibrillation was the most common toxicity leading to discontinuation. From real-world data, however, most patients developing atrial fibrillation required only temporary cessation of drug for medical rate control, followed by prompt reintroduction.49,50 Toxicities such as arthralgia and diarrhoea are not so much serious as annoying and may warrant dose reduction. Hence, new BTK inhibitors that reduce the risk of even minor toxicities that lead to treatment fatigue, can help ensure patients adhere to the long-term therapy needed for durable disease control.
Newer Bruton’s tyrosine kinase inhibitors
Zanubrutinib
As a non-selective BTK inhibitor, ibrutinib binds to other TEC family enzymes, epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR), human epidermal growth factor receptor 2/4 (HER2/4), B-lymphoid tyrosine kinase (BLK) and JAK3,51 predisposing to off-target toxicities unrelated to its antitumour effect. Zanubrutinib is not only a more potent BTK inhibitor than ibrutinib, but it is also more selective with less effect notably against EGFR,52 and therefore may have an improved toxicity profile. As it is a covalent BTK inhibitor and binds to the same cysteine-481 in the ATP pocket of BTK as ibrutinib, it is not expected that zanubrutinib would differ significantly from ibrutinib in efficacy.
Zanubrutinib received its first global drug approval for WM in Canada in 2021, founded on results from the ASPEN trial, a randomized phase III trial comparing ibrutinib with zanubrutinib.53 This trial was based on encouraging data from an early phase study of zanubrutinib monotherapy in various B-cell malignancies including WM.54,55 In this study, no dose-limiting toxicities were reported and the 160 mg twice-daily dosing was found to be optimal for sustained BTK occupancy. In the phase II expansion part of the study, 77 patients with WM (24 TN and 53 R/R) treated with either 160 mg twice daily or 320 mg daily, were able to achieve an MRR of 82.2%, VGPR of 45.2% and PFS of 80.5% at a median follow-up of 36 months.56 Furthermore, it was promising to see that selected toxicities such as atrial fibrillation (all grades 5.2%) and diarrhoea (all grades 19%) appeared less frequently than with ibrutinib.
The randomized phase III ASPEN trial was therefore designed to directly compare safety and efficacy of ibrutinib and zanubrutinib in TN or R/R WM patients with MYD88L265P.53 A third cohort of patients with MYD88WT were not randomized and all received zanubrutinib until progression.57 A total of 201 MYD88L265P patients were randomized, with the primary endpoint of CR or VGPR. No patients in either arm achieved a CR, but a non-statistically significant trend towards a higher VGPR rate with zanubrutinib (28%) compared with ibrutinib (19%) was reported (p=0.09). This trend was similar in both R/R and TN disease. At a median follow-up of 19.4 months, the median PFS was not reached in either arm. Therefore, this study demonstrated comparable efficacy between zanubrutinib and ibrutinib. However, it is worth noting that zanubrutinib appeared to be more rapid in onset of response, particularly in TN patients and those with CXCR4MUT.53 Furthermore, in the separate cohort of 26 MYD88WT patients who typically have dismal outcomes, zanubrutinib led to surprisingly good outcomes: ORR 81%, VGPR 27% and 18-month PFS 68%.57 This provides fodder for further investigation of zanubrutinib in this poor-risk group.
From the toxicity perspective, zanubrutinib appeared to be better tolerated than ibrutinib, with at least a 10% decrease in the incidence of all grades adverse events, including diarrhoea (21% versus 32%), atrial fibrillation (2% versus 15%), contusion (13% versus 24%), peripheral oedema (9% versus 19%), muscle spasms (10% versus 24%) and pneumonia (2% versus 12%).53 The exception was neutropenia, which was reported more frequently with zanubrutinib (all grades 29% versus 13%), but without excess infections. More patients in the ibrutinib group required dose reduction (24% versus 13%) or discontinued treatment (9% versus 4%) due to adverse events.
Overall, the ASPEN trial suggests that zanubrutinib may be better tolerated than ibrutinib, and until more confirmatory data emerge, it is reasonable to consider its use over ibrutinib in those patients at particular risk of toxicities. Alternatively, one may consider switching to zanubrutinib if intolerant but not progressing on ibrutinib. The potential for deeper and faster onset of response with zanubrutinib is intriguing and might be considered for patients with neuropathy, where symptomatic improvement is associated only with virtual elimination of circulating paraprotein. Again, studies confirming this approach are needed.
Acalabrutinib
Similarly to zanubrutinib, acalabrutinib is another covalent binding BTK inhibitor that binds to cysteine-481 for activity and has much more selectivity for BTK than ibrutinib.58 The ELEVATE-RR study was a head-to-head comparison of acalabrutinib and ibrutinib in CLL and was recently presented in abstract form at the 2021 American Society of Oncology meeting.59 Acalabrutinib was non-inferior to ibrutinib on the primary endpoint of PFS (hazard ratio 1.00) but demonstrated fewer adverse events and treatment discontinuations due to toxicity. When compared with ibrutinib, acalabrutinib led to significantly less atrial fibrillation (all grades 16% versus 9.4%) and hypertension (all grades 23.2% versus 9.4%) over the 41 months of follow-up. Though these data are promising in CLL, one cannot fully extrapolate to WM, where evidence supporting the efficacy and safety of acalabrutinib are largely restricted to a phase II single-arm study.60 In this study, 106 patients with either R/R WM (n=92) or TN disease (n=14) received acalabrutinib 100 mg twice daily until progression. Responses were achieved in 93% of all patients, with no CR and 9% VGPR at a median follow-up of 27 months. Fifty patients were evaluated for response by MYD88 mutational status. The MRR was 78% for patients with MYD88L265P and 57% for those with MYD88WT, which is similar to what one may expect with ibrutinib. In contrast to zanubrutinib, none of the 14 patients with MYD88WT achieved a VGPR. Once again, numbers are small, so confirmatory data are needed in this rare but high-risk subset of MYD88WT. Of note, the impact of CXCR4 mutations was not reported. Acalabrutinib has not yet been approved for the treatment of WM in Canada.
Tirabrutinib
Tirabrutinib is another highly potent and selective covalent-binding BTK inhibitor under investigation for WM. In a phase II single-arm study, 18 patients with TN and nine patients with R/R WM received oral tirabrutinib 480 mg daily.61 The MRR was 88.9% for all patients, with a median time to major response of 2 months. After a median follow-up of 6.5 months for TN patients and 8.3 months for R/R, all but one of the major responders were still in a major response. Similarly to ibrutinib, the MMR appeared to be higher for CXCR4WT (90.9%) than for CXCR4MUT (66.7%).
Most common toxicities with tirabrutinib were rash (44.4%), neutropenia (25.9%), leukopenia (22.2%) and stomatitis (14.8%), with cytopenias the most common grade 3–4 events.61 Encouragingly, bleeding occurred in only three patients (all grade 1) and there were no incidents of drug‐related atrial fibrillation or hypertension at short-term follow-up. Tirabrutinib therefore has a promising toxicity profile, but additional data will be needed before approval in Canada for WM.
Non-covalent Bruton’s tyrosine kinase inhibitors
The main mechanism of resistance to ibrutinib in WM is the acquisition of a BTK mutation at the C481 site.62 As this is also the binding site for covalent binding of BTK inhibitors such as acalabrutinib, zanubrutinib and tirabrutinib, these drugs are unlikely to be useful following failure of ibrutinib. Newer BTK inhibitors that do not bind to residue C481 are under investigation in various B-cell malignancies, including WM. One such agent, pirtobrutinib (previously LOXO-305), has demonstrated preliminary efficacy in patients with WM as part of a phase I/II trial of patients with advanced B-cell malignancies who received more than two prior lines of treatment.63 Among the 19 efficacy-evaluable patients with WM, the ORR was 68% including nine major responses (MRR 47%). Interestingly, the ORR was 69% even in patients with prior BTK inhibitor exposure/failure, with an MRR of 38%. In the larger CLL cohort, it is notable that despite 86% having previously failed BTK inhibitor therapy, the ORR was similar to that of BTK inhibitor-naïve patients, and responses were not impacted by discontinuation for BTK inhibitor progression or intolerance.64 Furthermore, the toxicity profile for pirtobrutinib was very favourable, with only fatigue (20%), diarrhoea (17%) and contusion (13%) occurring in at least 10% of patients. The results are promising in addressing the unmet needs of patients who are refractory to covalent-binding BTK inhibitors.
Another non-covalent BTK inhibitors, MK-1026 (formerly ARQ-531), is under investigation. It is believed to be less selective than pirtobrutinib, with significant activity on other kinases including SRC, extracellular signal-regulated kinases (ERK) and AKT.65 A phase Ib trial was completed, mainly in patients with CLL,66 and a phase II trial in various B-cell malignancies is ongoing. Additional non-covalent BTK inhibitors are in preclinical development and thus far have shown in vitro activity against wild-type BTK as well as C481-mutant BTK.67,68
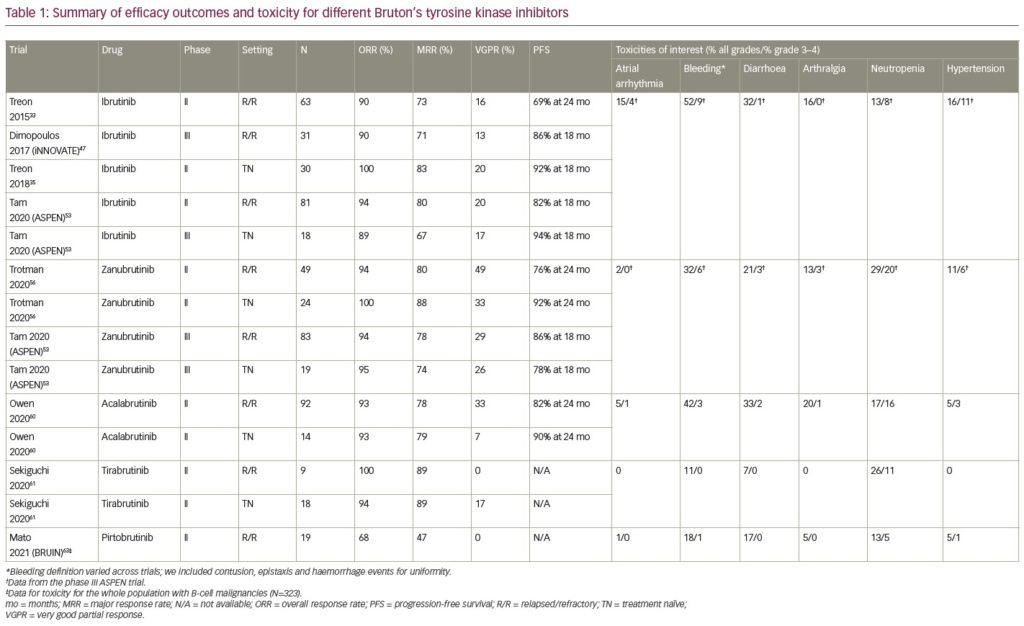
A summary of efficacy outcomes and toxicity for different BTK inhibitors is provided in Table 1.
Conclusion – Where do Bruton’s tyrosine kinase inhibitors fit in the Canadian treatment landscape
Ibrutinib, the first BTK inhibitor approved for use in WM, has changed the landscape of treatment in WM by demonstrating remarkable, durable responses coupled with a tolerable safety profile. However, when given as continuous monotherapy, even minor toxicities can lead to treatment fatigue. Therefore, newer, better-tolerated BTK inhibitors such as zanubrutinib and acalabrutinib are important breakthroughs in the treatment of WM. By improving drug adherence, they may lead to prolonged disease control. We have recently learned that MYD88 and CXCR4 genotyping can stratify patients for BTK inhibitor response, most importantly identifying those very poor responders with MYD88WT/CXCR4WT for whom traditional BR chemoimmunotherapy may still be preferred. BR is attractive not only for its impressive efficacy (PFS of 8 years or beyond in the frontline setting), but also for its finite 6-month course, affording a long drug-free period before next treatment, manageable safety profile and cost-effectiveness, the latter being of particular interest to publicly funded healthcare systems such as in Canada. A recent analysis of the Canadian cohort from the Alliance trial on patients with CLL, which compared BR with ibrutinib, showed that the total mean costs (over 2 years) were $192,615 with ibrutinib and $55,413 with BR.69 This cost differential was mainly due to the cost of ibrutinib itself, with the presumption that this cost differential would widen over time with ongoing ibrutinib therapy. A recent publication from WhiMSICAL (Waldenström’s Macroglobulinaemia Study Involving CART-WHEEL), a global registry of WM patient-derived data, supports the use of BR in the first-line setting, suggesting a longer time to next treatment compared with a BTK inhibitor.70
However, as we move towards greater access to testing of CXCR4, MYD88L265P and variants, selected BTK inhibitors may emerge as superior for specific genotypes, warranting their prioritized use, despite the added cost. Until then, BR remains a reasonable frontline standard of care in patients with WM. That said, for frail or elderly patients who may struggle with the toxicities of BR, or in the second-line setting where chemoimmunotherapy regimens fall short,71,72 ibrutinib has a clear role as standard of care. Here, the different BTK inhibitors may start to distinguish themselves, with one or another emerging superior in selected subgroups. It is indeed an exciting time for WM, with new drugs such as the BTK inhibitors under development on the basis of molecular mechanisms of disease and evaluated in studies specifically focusing on patients with WM.








