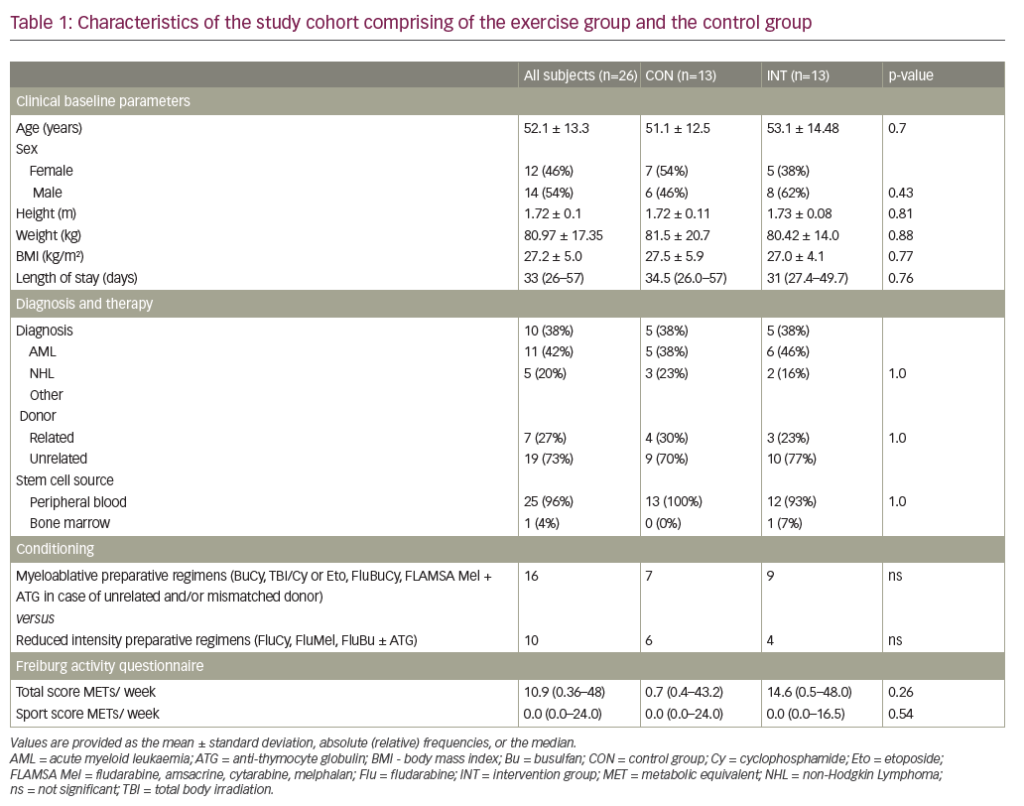
The chronic anaemia of beta-thalassemia major (b-thal) results from defective erythropoiesis secondary to a genetic defect in haemoglobin synthesis.1 Consequently, patients with
moderate-to-severe disease rely on blood transfusions to maintain an adequate level of haemoglobin.2 Over time, this causes an iron-overloaded state, in addition to the possible
blood-borne infections resulting from repeated transfusions.3 The widespread use of iron chelators has greatly improved the quality and lifespan of patients with b-thal and in recent years many advancements have been made in an attempt to reduce the dependency on blood transfusions for treatment. Some of the noteworthy lines of treatments that have received consideration in the last decade include gene therapy, allogeneic haematopoietic stem cell transplantation and pharmacological agents such as luspatercept.4
Luspatercept is a recombinant fusion protein with a modified extracellular domain of the human activin receptor type IIB, joined to the fragment crystallisable domain of human immunoglobulin G1, which attaches to a subset of transforming growth factor beta superfamily ligands, leading to complex downstream signalling pathways, resulting in increased erythropoiesis.5 The US Food and Drug Administration (FDA) first approved luspatercept for the treatment of anaemia in
transfusion-dependent adults with b-thal in the USA in November 2019,6 based on the results of the phase III BELIEVE trial (ClinicalTrials.gov Identifier: NCT02604433), which showed that the use of luspatercept resulted in a significant decrease in the need for packed red blood-cell (pRBC) transfusions.7
The large-scale double-blind trial was performed in over 60 centres across 15 countries and included pRBC transfusion-dependent patients with confirmed b-thal over the age of 18 years. The median age of the cohort was 30 years. These patients were randomised 2:1 to receive
1.0 mg/kg luspatercept subcutaneously, which was titrated up to 1.25 mg/kg, or placebo (n=224 and n=112, respectively) every 3 weeks for at least 48 weeks. All patients also continued to receive transfusions and iron chelation therapy. Of the patients in the treatment arm, 21.4% reached the study’s primary endpoint of ≥33% reduction in transfusion burden plus a reduction of ≥2 pRBC units during weeks 13–24, compared to a 12-week baseline period, versus 4.5% of patients in the placebo arm (odds ratio 5.79; p<0.0001). Furthermore, 70.5% of the patients in the treatment group achieved a ≥33% reduction in transfusion burden during any consecutive 12 weeks of treatment. In comparison, only 29.5% of patients in the placebo group achieved the same result (p<0.001).
Adverse events (AEs) were generally mild and tolerated, with grade 3 or higher AEs occurring 29.1% of patients treated with luspatercept and 15.6% of patients in the placebo group.
The most common grade 3 or higher AEs were anaemia (3.1% versus 0%), increased liver iron concentration (2.7% versus 0.9%) and hyperuricaemia (2.7% versus 0%) in the luspatercept and the placebo groups, respectively. Other commonly seen AEs in >5% of patients in the treatment group included bone pain, arthralgias, hypertension and dizziness. Most of these symptoms were controlled with medications, although permanent discontinuation of treatment because of an AE was required in 5.4% of patients treated with luspatercept because of arthralgias, back and bone pain. Dose reductions were required in 2.7% of patients, most frequently because of hypertension and headache.8 Thromboembolic events were seen in 3.6% of patients in the luspatercept group and 0.9% of patients in the placebo group. All of these events occurred in patients with a history of splenectomy as well as at least one more risk factor for thromboembolic events at baseline.7
By decreasing the transfusion burden, luspatercept presents an exciting option that could improve quality of life for patients with b-thal; however, the cost of the drug is substantially higher than the standard therapy and the need for continuous therapy would likely increase the cost of b-thal management.9 This is an important factor, as in developing countries in South Asia, the prevalence of the disease is fairly high due to the high genetic prevalence and the common practice of consanguineous marriages.10 Also, with novel treatment alternatives under development, such as gene therapy and stem cell transplant,11,12 which would potentially eliminate the need for erythropoiesis-stimulating agents, the role of luspatercept in the context of such therapies might be minimal. Therefore, despite all its merits, the cost of luspatercept can be a barrier to treatment for patients with b-thal in areas of high prevalence. Clinical trials investigating the long-term safety profile of the drug, as well as its use in non-transfusion dependent patients with b-thal minor, are currently ongoing (i.e., the BEYOND trial [ClinicalTrials.gov identifier: NCT03342404] and a study to evaluate long-term safety in subjects who have participated in other luspatercept clinical trials [ClinicalTrials.gov identifier: NCT04064060).8