Paroxysmal nocturnal haemoglobinuria (PNH) is a rare, chronic haematological illness. The condition presents on a background of bone marrow failure, and affected individuals can experience an array of symptoms due to chronic haemolysis, as well as episodes of acute haemolysis and thrombotic events.1 Diagnostic delays in PNH are common as not all patients experience classical symptoms, such as haemoglobinuria, at initial presentation.2 In 2007, the US Food and Drug Administration (FDA) approved the use of eculizumab for treating PNH. Eculizumab is a humanized monoclonal antibody that stops the formation of terminal complement, thereby preventing intravascular haemolysis.3 It effectively reduces morbidity and improves mortality to near-normal expectations.4–7
Other complement inhibitors have been developed for treating PNH, with the FDA approving ravulizumab in 2018 and, more recently, in May 2021, pegcetacoplan. This review describes the condition and its treatment, focusing on the rationale for the use of pegcetacoplan.
Disease pathophysiology
Most cases of PNH are due to a somatic mutation of the phosphatidylinositol glycan A (PIG-A) gene, which is one of over 25 genes required to form the glycophosphatidylinositol (GPI) anchor within the endoplasmic reticulum (ER).8–11 The GPI anchor is formed via a stepwise addition of sugar nucleotides and phospholipids within the ER, before the completed protein is transferred to the cell surface.12 The GPI anchor holds a wide variety of cell surface proteins that play important roles in signal transduction and the immune response.13,14 PIG-A mutations disrupt GPI production causing an absence of the GPI anchor and consequently of GPI-linked proteins. CD55 (decay accelerating factor) and CD59 (membrane inhibitor of reactive lysis) are GPI-linked proteins widely expressed on haematopoietic cells and are responsible for regulating complement. PNH red blood cells can be classified as either type II cells, with a reduced expression of CD55 and CD59, or as type III cells, with a complete absence of CD55 and CD59.15 Without expressing complement regulatory proteins, PNH red cells are lysed in the circulation (intravascular haemolysis). PNH platelets are not lysed but instead undergo conformational change, whereas PNH leucocytes become activated and release pro-inflammatory cytokines. These effects on PNH cells are implicated in the different symptoms experienced by patients with PNH.
PIG mutations in paroxysmal nocturnal haemoglobinuria
PIG-A is on the X chromosome and therefore mono-allelically expressed, so only a single mutation in the gene is needed to disrupt GPI formation. All the other genes involved in GPI biosynthesis are autosomal; for other genes to be causative would require both copies of the gene to be mutated. Mutations in other genes, PIG-M, PIG-B and PIG-T, have all been described.16–19 PIG-M has been identified in two unrelated consanguineous families. Affected individuals present in childhood with thrombosis and epilepsy.16 Four cases with a germline mutation in one PIG-T allele and a somatic mutation in the other PIG-T allele have also been reported.20 All four of these PIG-T-mutated cases experienced auto-inflammatory symptoms preceding the clinical symptoms of PNH. Langemeijer et al. described a rare familial form of PNH due to a homozygous mutation in PIG-B, with affected family members also experiencing unusual inflammatory symptoms, with inflammatory features commencing just before the diagnosis of PNH.17
Paroxysmal nocturnal haemoglobinuria development and classification
There is a strong association between PNH and bone marrow failure.21–25 It is likely that PNH only develops on a background of bone marrow failure, in an environment that allows the PNH clone to expand, as a PIG-A mutation in a haematopoietic stem cell on its own is not enough to cause PNH.26–28 PNH clones have been reported in over 50% of individuals with aplastic anaemia.28–32 However, it is not clear what causes the proportion of these PNH cells to increase in some cases.
In general, the more PNH blood cells are produced, the more likely PNH symptoms will manifest. A classification of PNH was devised in 2005 by Parker et al. on behalf of the International PNH Interest Group with the following categories: classic PNH, PNH occurring in the presence of another specified bone marrow disorder, and subclinical PNH.21 Patients with classic PNH have evidence of intravascular haemolysis but not of another bone marrow disorder, whilst patients with PNH in the presence of another bone marrow disorder have intravascular haemolysis, either with a history of, or with another defined bone marrow condition. Subclinical PNH is where a small proportion of PNH cells are present but there is no haemolysis. In clinical practice, there is a significant overlap between the first two categories.
Laboratory diagnosis of paroxysmal nocturnal haemoglobinuria
Flow cytometric detection is the gold standard method for diagnosing and monitoring PNH. This technique allows evaluation of PNH neutrophils and erythrocytes down to a level of 0.01%.33 The International Clinical Cytometry Society recommends gating antibodies CD45, CD15 and CD64 for assessing neutrophils and monocytes, combined with fluorescent-labelled proaerolysin (FLAER), CD24, CD14 and CD157 for detecting PNH cells.33 FLAER can be used to assess neutrophils and monocytes, as it binds directly to the GPI anchor, which is more specific and sensitive to analyse, rather than to a single GPI-linked protein, of which there are multiple, on the surface of these cells. Assessment of erythrocytes is also recommended using CD235a for gating and CD59 for detecting PNH cells.
It is essential to assess PNH clones in the white cell lineage to accurately determine the size of the PNH clone. The proportion of PNH cells identified in erythrocytes may be lower than in white cells, due to intravascular haemolysis and red cell transfusions required to treat anaemia. Assessing erythrocytes may provide useful clinical information, including the percentage of type II and type III cell populations, and adding C3d to this assay enables rapid flow cytometric assessment of extravascular haemolysis resulting from inhibiting complement at complement protein C5.34
Clinical features of paroxysmal nocturnal haemoglobinuria
Chronic haemolysis is the underlying cause of the complications observed in PNH. Patients experience variable signs and symptoms, including haemoglobinuria, fatigue, dysphagia, abdominal and chest pain, renal impairment, erectile dysfunction and an increased incidence of thrombosis.35
Thrombosis
Prior to complement inhibition, thrombosis was the main cause of mortality in PNH. It may also be the presenting event leading to a diagnosis of PNH. Around 29–40% of patients will develop thromboses during the course of the disease.1,36–39 It is essential to diagnose and promptly treat patients presenting with thrombosis to reduce clot propagation; anticoagulation alone is insufficient in managing a PNH thrombotic event. Thrombotic events commonly manifest as pulmonary emboli and deep vein thromboses, but there is an increase in clot formation in unusual sites, such as the liver (Budd–Chiari syndrome), abdomen and cerebral veins.1,40 Fifteen percent of events are arterial.1 Thrombotic events occur due to a combination of factors, including intravascular haemolysis, the release of free haemoglobin, platelet and leucocyte activation, and endothelial activation.39
Other disease complications
Renal impairment is present in around two-thirds of patients with PNH.41 In most cases, this is mild in nature with stage 1 and 2 chronic kidney disease, but around 20% of cases are stage 4 or 5. Renal disease occurs as a result of haemosiderin deposition in the renal tubules, vasoconstriction of blood vessels, and microthrombi.42–44 Nitric oxide uptake also disrupts smooth muscle function, leading to symptoms including dysphagia, abdominal pain, breathlessness and erectile dysfunction.45
Treatment of paroxysmal nocturnal haemoglobinuria
General supportive measures in paroxysmal nocturnal haemoglobinuria
A diagnosis of PNH can be confusing for patients. Accessing specialist support can be difficult, but both good disease information and patient support groups are helpful. Not all patients with PNH require pharmacological management. Patients with smaller PNH clones and those without indications for treatment should be managed proactively with folic acid and close monitoring for symptoms, PNH clone changes and the development of other bone marrow failure disorders.
Patients with larger PNH clones who are not indicated for complement inhibition should be considered for prophylactic anticoagulation where safe to do so. In countries where complement inhibition is not available, anticoagulation should also be considered.
Inhibition of complement in PNH
Complement inhibition is generally indicated in patients with symptomatic intravascular haemolysis, PNH-related thrombosis (clinical emergency) or PNH-related longer-term complications, such as renal failure and pulmonary hypertension. Most centres also advocate the use of eculizumab during pregnancy (unlicensed use).
Complement inhibition is not available in many countries worldwide, due to the high cost of the treatment, thus creating a disparity, which, in time, should be addressed by the increasingly competitive nature of the treatment landscape.
Inhibition of complement at C5
Eculizumab and ravulizumab block terminal complement formation by binding to the complement protein C5 and preventing its cleavage to C5a, an anaphylatoxin, and C5b, which forms part of the membrane attack complex (Figure 1). These therapies can block intravascular haemolysis, thereby reducing disease morbidity and mortality.4–7,46,47
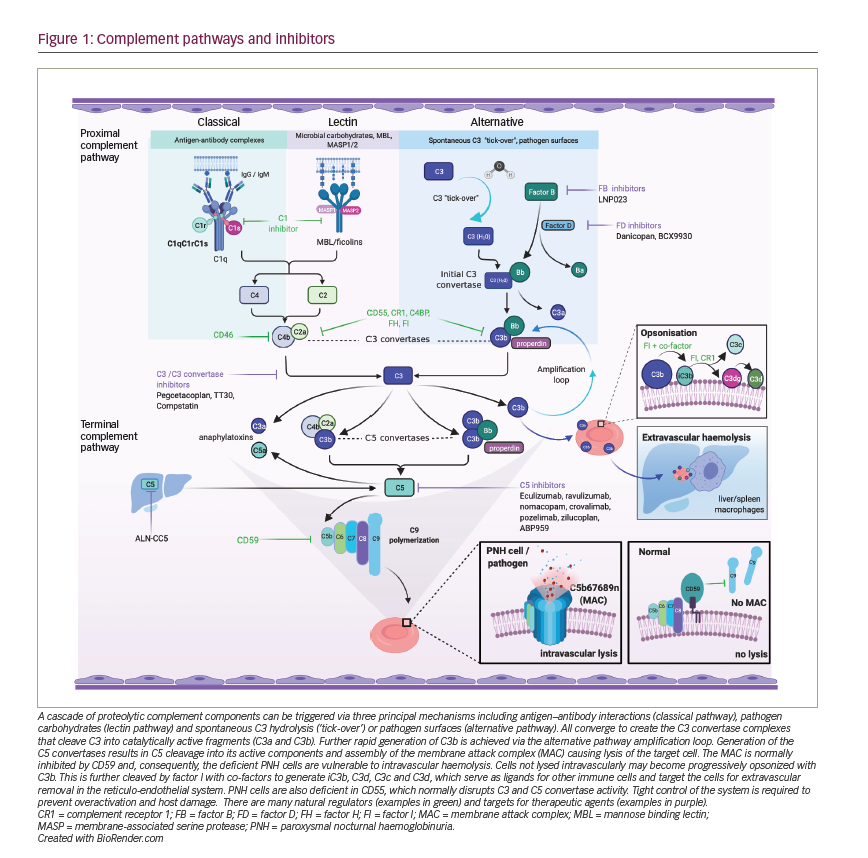
Eculizumab was approved by the FDA and the European Medicines Agency for the treatment of PNH in 2007 and has been widely used since its approval. Initially, 600 mg is given weekly for the first 4 weeks, and then 900 mg every 2 weeks thereafter.4–6 It has also been used in pregnancy with good maternal and foetal outcomes.48
The FDA approved ravulizumab for use in PNH in 2018. Ravulizumab, which has a half-life four times that of eculizumab, is administered according to weight as an initial loading dose, followed by a maintenance dose 2 weeks later, and then every 8 weeks. The loading dose is 2,400 mg for patients weighing 40–60 kg, 2,700 mg for those weighing 60–100 kg and 3,000 mg for those weighing over 100 kg. The maintenance dose for these weight ranges is 3,000 mg, 3,300 mg and 3,600 mg, respectively. Phase III trials have demonstrated non-inferiority to eculizumab both in untreated patients and in those who were stable on eculizumab.46,47
Complement-driven extravascular haemolysis
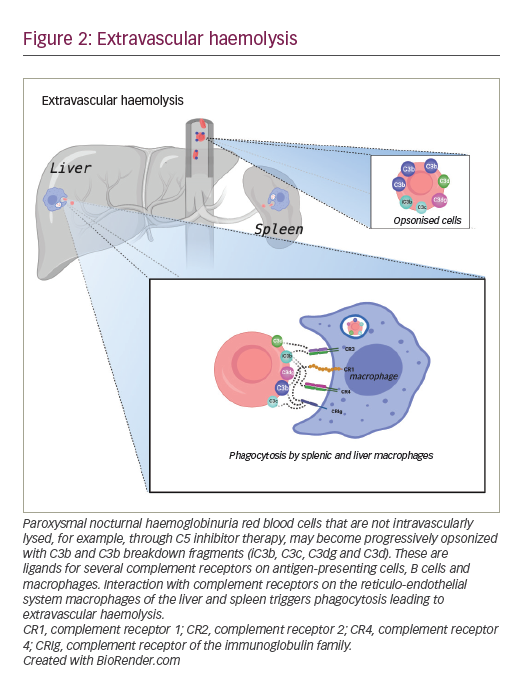
Whilst complement inhibition targeting the complement protein C5 is highly effective in preventing intravascular haemolysis and its consequences, the use of these therapies may lead to an increase in extravascular haemolysis of PNH erythrocytes. Extravascular haemolysis in patients receiving C5 inhibitors is driven by C3 fragments opsonizing the PNH red cells, marking them for destruction within the reticulo-endothelial system (Figure 2).34,49 Although anaemia in PNH can be multifactorial with aplasia also being implicated, extravascular haemolysis is experienced by most patients on C5 inhibitors, causing anaemia and, in some cases, necessitating blood transfusions. In an analysis of 141 patients maintained on eculizumab for at least 13 months, 72% were anaemic with 36% of these requiring at least one transfusion per year.50
Inhibition of complement at C3: pegcetacoplan
Pegcetacoplan is a new class of complement inhibitor. It is a pegylated synthetic peptide conjugated to a polyethylene glycol polymer that binds to the complement protein C3 preventing its cleavage to C3a and C3b. Pegcetacoplan is a self-administered, twice-weekly subcutaneous injection (1,080 mg), and prevents both intravascular and extravascular haemolysis by preventing C3 opsonization of PNH red blood cells (Figure 3).
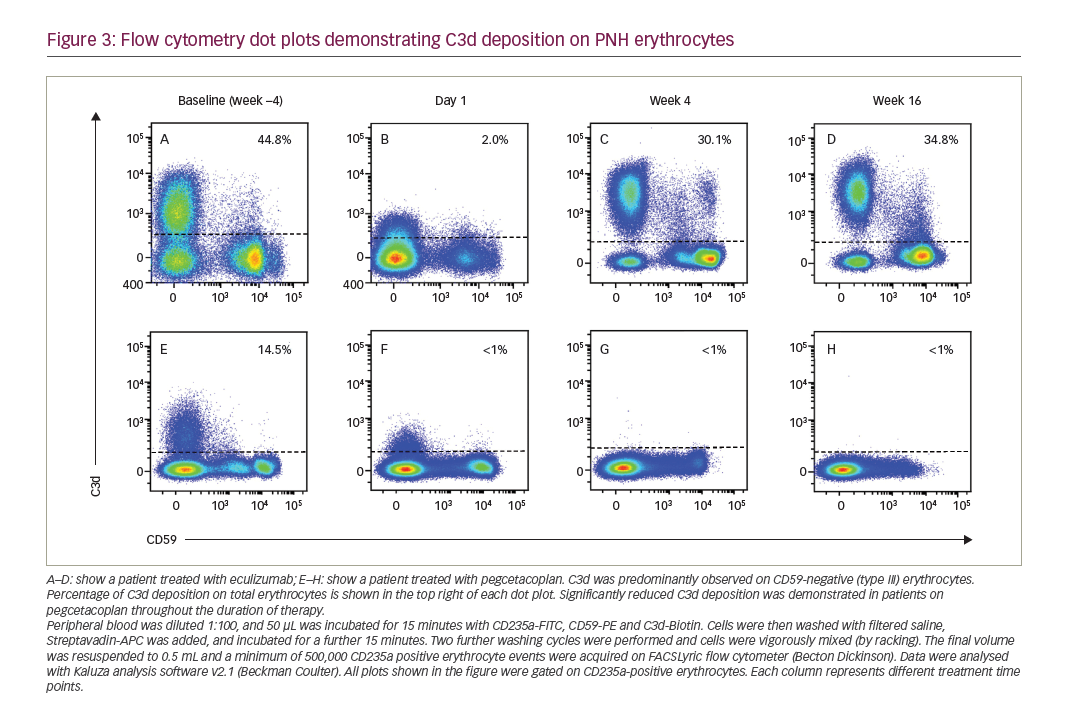
De Castro et al. assessed the safety, efficacy and pharmacokinetics of pegcetacoplan in a phase I trial of patients who continued to experience anaemia despite eculizumab therapy.51 Nine patients were enrolled, with four completing the study and continuing on pegcetacoplan in an extension study. Pegcetacoplan was well tolerated, with increases in haemoglobin and decreases in bilirubin and reticulocyte levels observed.
The key 16-week trial data are from the phase III, open-label clinical trial (Pegasus trial; ClinicalTrials.gov identifier NCT03500549), which evaluated the efficacy and safety of pegcetacoplan compared with eculizumab.52 Adult patients who were diagnosed with PNH by flow cytometry and who had a haemoglobin level <10.5 g/dL whilst receiving stable doses of eculizumab for at least 3 months were eligible for the study. The trial was divided into three distinct parts, with a 4-week ‘run-in’ period where patients received their previous eculizumab therapy as well as twice-weekly pegcetacoplan (1,080 mg per injection). After the run-in period, patients were randomized 1:1 to receive either eculizumab or pegcetacoplan for 16 weeks, followed by a 32-week open-label extension where all were treated with pegcetacoplan. During the open-label extension, the patients from the eculizumab group were treated with eculizumab and pegcetacoplan for the initial 4 weeks, followed by 28 weeks of pegcetacoplan monotherapy. The 1:1 randomization was stratified for platelet count higher or lower than 100 × 109/L, and greater or fewer than four transfusions in the preceding 12 months prior to enrolment in the trial.
The primary endpoint of the trial was the change in haemoglobin level from baseline to week 16. Secondary endpoints measured at week 16 included the proportion of patients who were transfusion independent, the change in reticulocyte and lactate dehydrogenase (LDH) levels and changes in the functional assessment of chronic illness therapy-fatigue (FACIT-F) score. Eighty patients (39 female, 41 male) were treated in the study, with 41 in the pegcetacoplan arm and 39 in the eculizumab arm. Three of those in the pegcetacoplan arm discontinued the study due to experiencing breakthrough haemolysis (BTH). The characteristics of the patients in the two arms were largely similar, including patient age, body mass index, prior history of aplastic anaemia, haemoglobin, platelet count, LDH, FACIT-F score and median duration on eculizumab. Twenty-four (30%) patients were receiving higher than standard doses of eculizumab at the start of the trial.
The primary endpoint was achieved, with a mean increase in haemoglobin in the pegcetacoplan arm of 2.37 g/dL compared with a reduction in the eculizumab arm of 1.47 g/dL at 16 weeks. Regarding the main secondary endpoints, significantly more patients were transfusion independent in the pegcetacoplan arm (35/41, 85%) when compared with the eculizumab arm (6/39, 15%) over the 16-week period. A clinically significant improvement in FACIT-F scores at week 16 was also observed in those receiving pegcetacoplan, with a 9.2 point increase compared with a 2.7 point decrease in those receiving eculizumab. Non-inferiority of pegcetacoplan was seen in absolute reticulocyte count levels, which decreased on pegcetacoplan and increased slightly on eculizumab (-136 ± 7 × 109/L versus 28±12 × 109/L, respectively). Non-inferiority was not seen in the mean change in LDH from baseline (15 ± 43 U/L with pegcetacoplan compared with 10 ± 71 U/L with eculizumab). More patients’ blood values became normal on pegcetacoplan compared with eculizumab; haemoglobin (34% versus 0%), LDH (71% versus 15%), reticulocytes (78% versus 3%) and bilirubin (63% versus 8%).
Adverse events occurred in a similar proportion of patients on pegcetacoplan as on eculizumab. Roughly equal levels of infections were seen, occurring in 29% on pegcetacoplan and 26% on eculizumab. No meningococcal or pyogenic infections were observed during the trial; however, follow-up was very short. Episodes of BTH occurred in four patients (10%) on pegcetacoplan and in nine (23%) on eculizumab. The other common adverse events on pegcetacoplan were injection site reactions (37%) and diarrhoea (22%). Pegcetacoplan has also been approved by the FDA for treatment-naïve patients with PNH who require complement inhibition.
Managing breakthrough haemolysis on proximal complement inhibitors
Episodes of BTH in patients treated with proximal complement inhibitors are potentially difficult to manage. Due to improvements in haemoglobin levels and consequently the proportion of PNH red cells, a stimulating event, such as infection, can result in brisk haemolysis and a rapid drop in haemoglobin. Whilst the degree of BTH can be marked, an additional dose of a C5 inhibitor can stop the haemolytic episode. Within the Pegasus trial, four patients on pegcetacoplan experienced BTH in the 16-week period.52 Whilst nine patients on eculizumab experienced breakthrough in the first 16 weeks, these events could be managed with additional C5 inhibition (outside of the clinical trial setting).
A strategy for managing BTH events on all proximal complement inhibitors, not just pegcetacoplan, is essential. This strategy should include good patient education, particularly for patients who may have experienced BTH on C5 inhibition, as symptoms can be different and haemolysis can be brisk. How to manage these events pharmacologically can be debated; for patients with suboptimal LDH control (>2 × upper limit of normal), the dose of pegcetacoplan can be increased to 1,080 mg every 3 days; however, this may not be sufficient, particularly during an acute haemolytic breakthrough, and an alternative treatment might be required, such as a C5 inhibitor.
Managing infection risk on complement inhibition
Individuals with a hereditary deficiency of C5 are unable to initiate an immune response to meningococcal infection, and complement inhibition provides a similar risk. Educating patients and their families is essential, as is vaccinating against meningococcal serotypes ACWY and B. Some centres advocate the monitoring of antibody titres to review and optimize the immune response, particularly to the ACWY vaccine (it is not possible to assess titres for serotype B due to C5 inhibition interfering with the assay), and providing meningitis B boosters every 5 years.53 Proximal complement inhibition raises the possibility of increased risk of infection with other encapsulated organisms, and thus it is advised that patients are vaccinated against Haemophilus and pneumococci alongside meningococcal vaccinations. Some centres also promote the use of prophylactic antibiotics and a ‘back-up’ emergency provision with ciprofloxacin, as well as prompt medical reviews if pyrexial.
Other proximal complement inhibitors
Whilst not the main discussion for this article, other proximal complement inhibitors are in clinical trials or early stages of development, including targeting factor B and factor D.54,55
How to choose the treatment option most suitable for your patient
Where possible, all patients with PNH should be offered entry into a clinical trial. For those commencing approved therapies, which treatment option to choose for your patient may become more complex. Current licensed treatments in the USA include eculizumab, ravulizumab and pegcetacoplan.
Data from the Pegasus trial support the use of pegcetacoplan in those with significant extravascular haemolysis on a C5 inhibitor. However, for those patients well controlled on C5 inhibitors, whilst the option to change to pegcetacoplan should be considered, there are merits in remaining on a C5 inhibitor. Data on pegcetacoplan in PNH are still in their infancy in terms of longer-term complications and thrombotic events. In individuals with a complicated thrombotic history, or those presenting with thrombosis as their diagnostic event for PNH, it is likely preferable to use C5 inhibition, at least initially.
Thrombocytopaenia, when severe, can mean pegcetacoplan is not suitable, as pegcetacoplan is administered subcutaneously. In women with PNH considering pregnancy, eculizumab would be the preferred treatment until more data are available in this setting.48
Conclusion
The treatment of PNH has been revolutionized by complement inhibition. Prior to eculizumab, patients were managed with supportive care and, in some cases, allogeneic bone marrow transplantation.1 By preventing terminal complement formation, eculizumab and ravulizumab reduce the complications of chronic intravascular haemolysis, thereby improving patient outcomes and quality of life.4-6,46,47 Despite this improvement, over 70% of patients on eculizumab remain anaemic, with some requiring transfusions due to extravascular haemolysis.50
Pegcetacoplan, by blocking complement earlier in the complement pathway, prevents intravascular haemolysis without causing an increase in extravascular haemolysis.52 This therapy, like C5 inhibitors, prevents the unwanted effects of intravascular haemolysis, but when compared with eculizumab, improves haemoglobin levels and, consequently, FACIT-F scores.
Whilst pegcetacoplan is a promising therapy for treating PNH, there is still far more clinical experience in C5 complement inhibitors. How to manage BTH, and how to manage patients presenting with a thrombosis or with complicated thromboses remain issues about which we will continue to learn and gather answers. Concerns regarding infection risk, in particular the increased risk of meningococcal infection, should be proactively managed.
The treatment landscape for PNH has changed significantly with newer therapies becoming available. The options for patients in a few years’ time may allow for an individualized approach to treatment. With oral, subcutaneous and long-acting intravenous options possible, we hope that pricing structure will be improved, widening access to patients worldwide who have indications for complement inhibition.
Pegcetacoplan is the first proximal complement inhibitor to be approved for use in PNH by the FDA. In patients who have ongoing anaemia due to extravascular haemolysis on C5 inhibition, pegcetacoplan may be a better therapy.