Multiple myeloma (MM) is the second most common haematological malignancy, with upwards of 35,000 diagnoses in the USA each year.1,2 It remains a leading cause of blood cancer–related mortality worldwide, and although therapeutic advances have allowed for significant improvements in the median overall survival,3,4 the majority of patients still experience cycles of relapse that are eventually fatal.5 While patients with MM are living longer, a subgroup with high-risk disease at diagnosis still does poorly, with a median overall survival of nearly 3 years.6 Outcomes are also dismal in patients with disease that is refractory to the major modern therapies including thalidomide analogues, proteosome inhibitors and anti-CD38 monoclonal antibodies.7 Together, these unmet needs have driven the development of immunotherapy for relapsed and refractory multiple myeloma (RRMM).
Toward this end, immune cell redirecting therapies were developed following the discovery of suitable MM cell-surface markers and novel drug engineering technologies. Chimeric antigen receptor (CAR)-T cells showed robust responses in RRMM compared with B cell maturation antigen (BCMA), leading to the approval of cellular therapies for MM – idecabtagene vicleucel in 2021 and ciltacabtagene autoleucel in 2022.8–11 Available data on idecabtagene vicleucel and ciltacabtagene autoleucel in RRMM, however, suggest that most patients will experience disease progression after initial response.9,11 Moreover, the need for patient-specific manufacturing and access to specialized cellular therapy centres continue to limit CAR T cell access for patients with rapid disease progression or in under-served areas.
Successful adaptive immune redirection has also been shown with bispecific antibodies (BsAbs) against various established and emerging MM cell targets. This review follows the US Food and Drug Administration (FDA) approval of teclistamab – the first-in-kind commercially available BsAb for RRMM.12 Teclistamab is a T cell-redirecting antibody that targets CD3 on the surface of T cells and BCMA expressed on the surface of myeloma cells (BCMA×CD3). It was approved for use as monotherapy in 2022 based on data from the MajesTEC-1 trial published in June 2022.13 Teclistamab and similar agents in development enable T cell redirection with similar potency to CAR T cells but without the delay required for patient-specific manufacturing.
In this article, we review the mechanism of action of this BCMA×CD3 antibody, the clinical data supporting the approval of teclistamab and its impact on the treatment paradigms for RRMM.
Mechanisms of action
Overview of B cell maturation antigen as a target
BCMA, also known as TNFRSF-17 or CD269, is a small transmembrane protein member of the tumour necrosis factor (TNF) receptor superfamily. It is selectively induced during plasma cell (PC) differentiation,14 with transcription and cell-surface expression concentrated in subsets of mature B cells, PCs and plasmacytoid dendritic cells.15,16 Select datasets have also suggested low-level transcription on neurons and astrocytes,17 though other studies have not supported this association.18,19
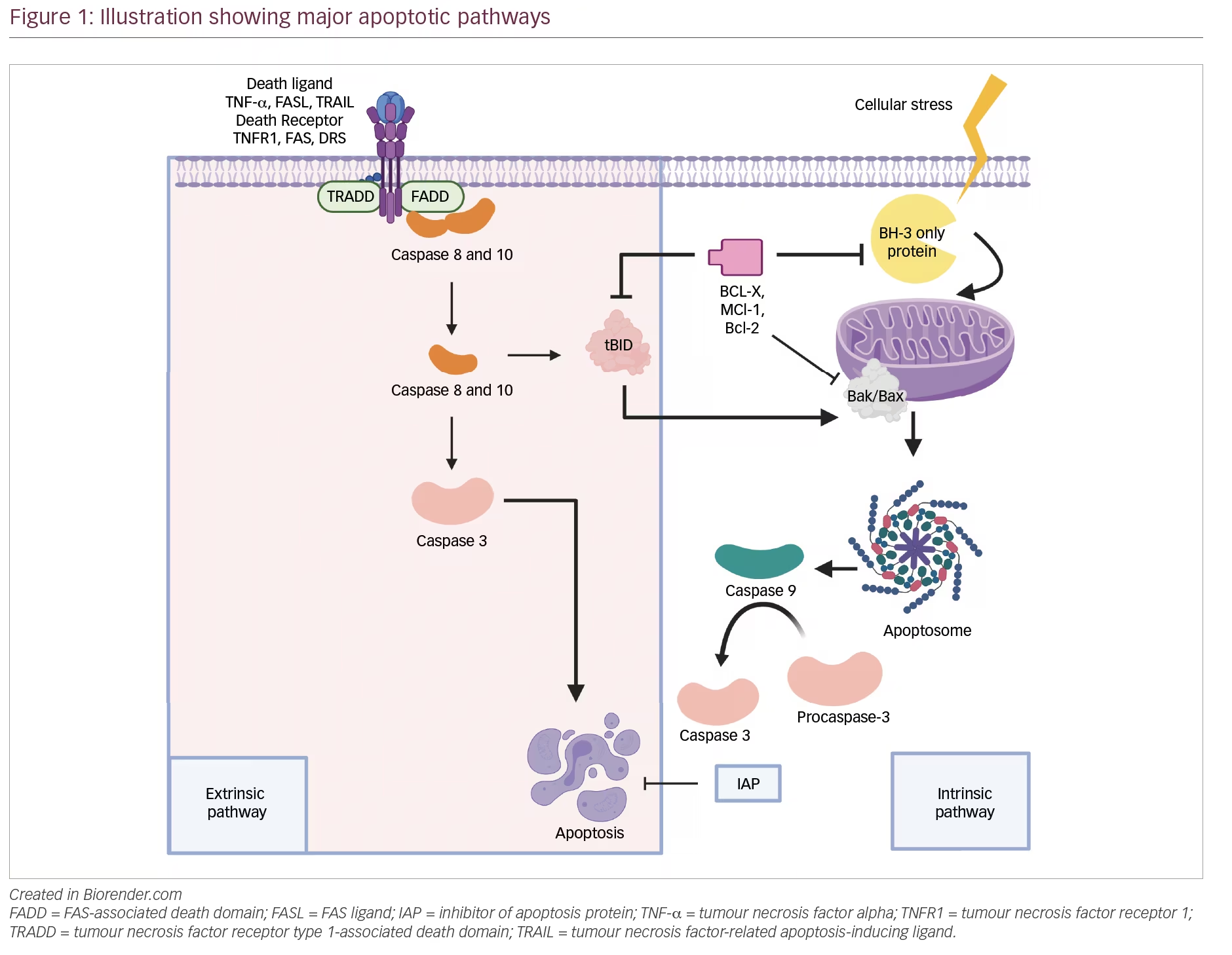
BCMA has two ligands, BAFF/BLys and APRIL, which are integral to maintaining bone marrow PC survival and homeostasis (as reviewed in Eckhert et al. and Romano et al.).20,21 Stimulation with APRIL or BAFF activates the NF-κB, AKT and mitogen-activated protein kinase (MAPK)8/c-Jun N-terminal kinase (JNK) signalling cascades, resulting in the upregulation of anti-apoptotic proteins,22 adhesion molecules, cell-cycle regulators, angiogenesis factors, immunosuppressive molecules and inflammatory cytokines (Figure 1A).23–26 Both ligands also bind to transmembrane activator calcium modulator and cyclophilin ligand interactor (TACI), a larger homologue receptor of the TNF receptor superfamily, and mediate PC differentiation and T cell-independent immunoglobulin (Ig) isotype switching.27 The role of BCMA activation, however, is restricted to maintaining PCs and antigen presentation by B cells.26 BCMA has a soluble form derived from the cleavage of the membrane BCMA mediated by γ-secretase.28 This soluble form has shown to sequester BAFF and mediate immunosuppression by preventing normal B cell and PC development.29
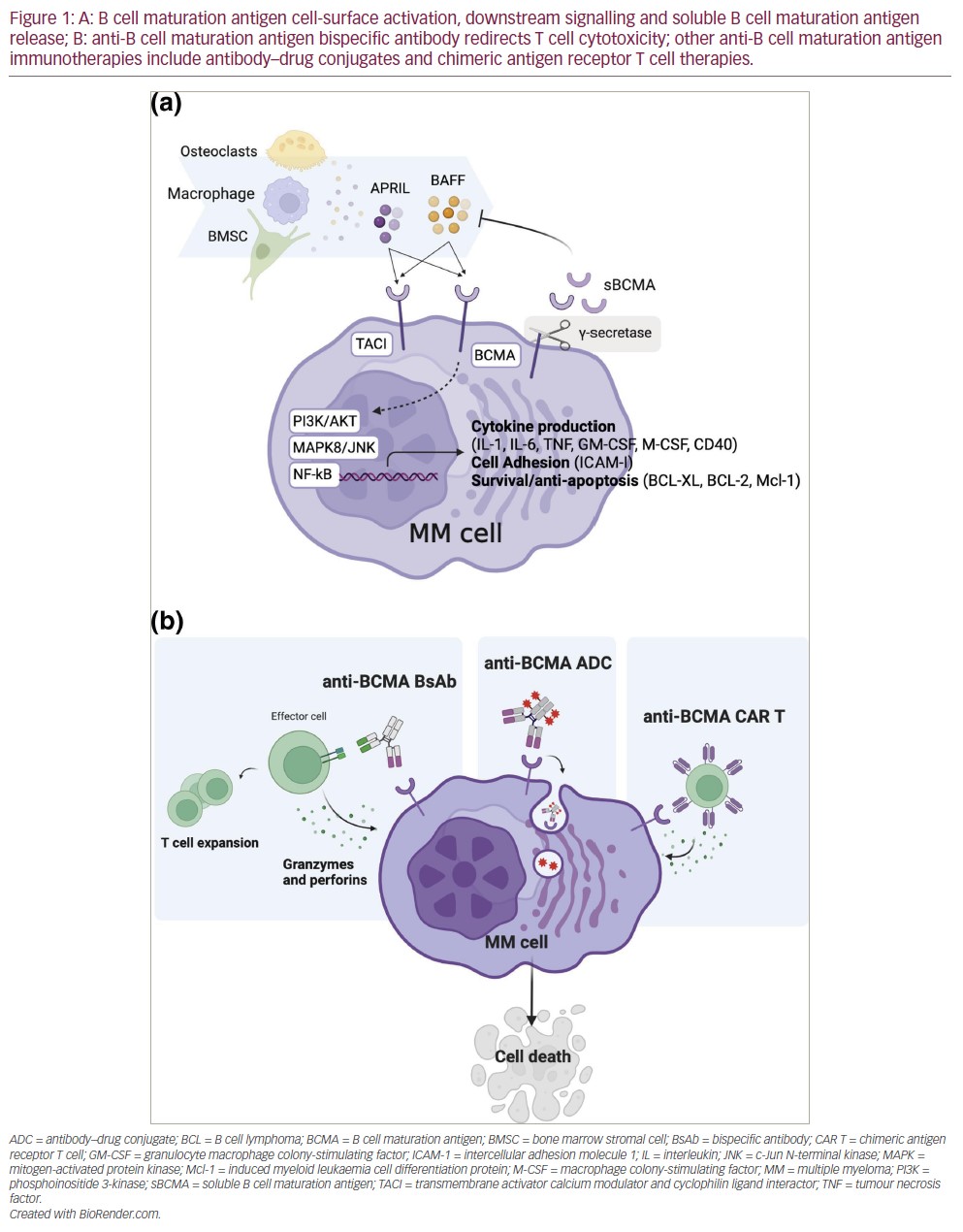
BCMA is overexpressed in MM,30 and NF-κB overactivation is a hallmark of MM tumourigenesis.31 In patients with MM, soluble BCMA is also significantly elevated compared with healthy individuals, and higher soluble BCMA (sBCMA) levels correlate with immune paresis, disease burden and adverse outcomes.28,32 Preclinical studies demonstrated that antibodies with ligand–blocking activity could promote cytotoxicity in MM cell lines as naked antibodies or as antibody–drug conjugates by inhibiting APRIL-dependent activation of NF-κB in a dose-dependent manner in vitro.33 BCMA was later validated as a suitable CAR T target.15 Altogether, these studies paved the way for the development and clinical investigation of anti-BCMA antibody–drug conjugates, CAR T and now BsAb therapies against RRMM (Figure 1B).
Bispecific antibody therapy development for relapsed and refractory multiple myeloma
BsAbs are T cell–engaging (TCE) therapies designed to redirect T cell cytotoxicity towards cell-surface tumour antigens (Figure 1B). TCE therapies have been developed in various formats, including bispecific IgG-like antibodies or shorter molecules comprising two linked antigen-binding domains configured as single-chain variable fragments, such as bispecific T cell engagers (BiTEs).34 BsAbs were originally proposed in the early 1960s but only developed35 and studied clinically36 years later. The favourable clinical efficacy of the blinatumomab (CD19×CD3 BiTE in relapsed B-cell acute lymphoblastic leukaemia) published in 201737 generated interest in BsAb development for other haematologic malignancies.34
An anti-BCMA BiTE construct, the BCMA×CD3 AMG420, yielded encouraging preclinical38 and clinical activity against RRMM.39 The need for a 4–week continuous infusion due to the short half-life of BiTEs shifted the focus onto longer half-life, full-length IgG molecules.40 Several BCMA-targeting BsAbs have since been developed and tested in various clinical settings and combinations, and are reviewed by Moreau and Touzeau.41 Of these, teclistamab has been studied in the MajesTEC trial series and will be further described in the following section.
BsAbs against other emerging targets in RRMM are also being investigated. Talquetamab, a G protein-coupled receptor class C, group 5, member D (GPRC5D)×CD3 BsAb, showed promising tolerability and efficacy in a phase 1 trial (MonumenTAL-1)42,43 and is also being studied in combination with daratumumab (TRIMM-2,44 MonumenTAL-345) and several other agents (MonumenTAL-2).46 Cevostamab, an Fc receptor-homologue 5 (FCHR5)×CD3 BsAb, has also demonstrated safety and promising efficacy in an on–going phase 1 study (ClinicalTrials.gov identifier: NCT03275103).47 This emergence of non-BCMA TCE targets is likely to introduce additional options and complexity in the care of heavily refractory patients.
Teclistamab in relapsed and refractory multiple myeloma
Clinical investigation
Teclistamab (also known as JNJ-64007957, Ab-957 and JNJ-7957) is a humanized IgG4-proline, alanine, alanine (IgG4-PAA) bispecific DuoBody® antibody (Genmab, Copenhagen, Denmark), whose in vitro efficacy was first shown in 2016.48,49 The first in–human trial with teclistamab, MajesTEC-1, was an open-label, single-arm phase 1/2 (phase 1 ClinicalTrials.gov identifier: NCT03145181; phase 2 ClinicalTrials.gov identifier: NCT03145181) trial that evaluated intravenous (phase 1) and subcutaneous (phase 1 and 2) administration.1,13,50,51 Participants were refractory to thalidomide analogues and proteosome inhibitors; most patients (93%) had also progressed on an anti-CD38 agent.51 Doses ranging from 0.3 µg/kg to 3000 µg/kg were evaluated.51 At most levels, a step-up dosing approach was used, in which 1–3 smaller quantities were administered over several days prior to the first full dose, with the intent of more gradually activating T cells and reducing the risk of severe cytokine release syndrome (CRS). Clinical responses were observed beginning at 38.4 µg/kg. No maximum tolerated dose was identified, and a recommended phase 2 dose of 1.5 mg/kg weekly, administered as a subcutaneous injection, was determined based on the combined safety, efficacy, pharmacokinetic and pharmacodynamic profiles of teclistamab. In total, 165 patients enrolled in phase 1 and 2 of the MajesTEC-1 trial received teclistamab at the recommended phase 2 dose. Patients received two step-up doses of 0.06 mg/kg and 0.3 mg/kg, which were separated by 2–4 days and were completed 2–4 days before the first full teclistamab dose was administered. Patients with confirmed partial response or better were permitted to switch to 2-weekly dosing.13,52
With a median follow-up of 14.1 months, the overall response rate was 63.0%, with 65 patients (39.4%) having a complete response or better. A total of 44 patients (26.7%) had no minimal residual disease; the negativity rate of minimal residual disease among the patients with a complete response or better was 46.0%. Responses occurred rapidly, with a median of 1.2 months until first response.
The Kaplan–Meier estimate of maintenance of response for at least 12 months was 68.5% (95% confidence interval [CI] 57.7–77.1). The median duration of response was 18.4 months (95% CI 14.9–not estimable). The median progression-free survival was 11.3 months (95% CI 8.8–17.1).
Safety and infection risk13
As with CAR T cells, CRS and immune effector cell-associated neurotoxicity syndrome (ICANS) are important adverse effects of teclistamab and occurred in 72.1% and 3.0% of patients, respectively. These risks are confined to the initial doses and typically occur 2 or 3 days following the inciting dose. Inpatient monitoring at the time of the two step-up dose and first full dose of teclistamab is recommended in the FDA prescribing information to enable the prompt management of CRS.53 Roughly half of the patients who experienced CRS, or 36% of phase 2 participants, received tocilizumab; 8.5% of subjects received corticosteroids for CRS or ICANS management, thus warranting the availability of anti-interleukin-6 agent upon therapy initiation. Patients who respond to teclistamab are not at an on–going risk of CRS or ICANS with long-term dosing. If therapy is interrupted for longer than a month, however, repeat step–up dosing is advised.54
Infections were major adverse events in the MajesTEC-1 trial and occurred throughout the therapy.50 A total of 126 patients (76.4%) reported at least one infectious event, with 74 (44.8%) experiencing grade 3 or 4 infections. Notably, coronavirus disease 2019 (COVID-19) was frequent and led to on-study mortality in 12 of the 165 participants, though many of the fatal cases occurred in the early phase of the pandemic. Other viral (cytomegalovirus, JC virus), bacterial and fungal events were also reported. Six (3.6%) patients developed Pneumocystis jirovecii pneumonia. Among the 19 on-study deaths due to adverse events, 14 were attributed to infection (12 due to COVID-19, one due to progressive multifocal leukoencephalopathy from JC virus infection, and one due to streptococcal pneumonia). Hypogammaglobulinaemia developed in 74.5% of patients (essentially all patients who responded to therapy and received long-term teclistamab), which likely contributed to the risk of infection. The FDA prescribing information recommends varicella zoster virus prophylaxis.53 Additional prophylaxis against Pneumocystis jirovecii pneumonia, and Ig replacement therapy to maintain IgG levels >400 mg/dL, should be considered.54 In our view, these measures are essential considering the magnitude of infection risk. Interventions to protect against COVID-19 are also prudent, including vaccination and proactive use of anti-virals when symptomatic infections develop. Though patients on teclistamab are not expected to mount antibody responses to vaccination, vaccine-induced T cell responses have been observed and may be clinically protective.55
Haematologic toxicity is commonly observed. Grade 3 or 4 neutropenia, anaemia and thrombocytopenia were reported in 64.2%, 37.0% and 21.2% of trial participants, respectively. Of the 117 patients in whom neutropenia developed, 91 received granulocyte colony-stimulating factor therapy at the investigator’s discretion.13,50,51 In our experience, neutropenia with teclistamab is idiosyncratic, occurring intermittently throughout treatment, and can be successfully managed with occasional dose holding and the administration of filgrastim.
Efficacy correlations and drug–drug synergy
Various baseline immune, tumour and clinical factors are associated with the clinical efficacy of teclistamab. 52 In MajesTEC-1, response rates and progression-free survival correlated positively with recipient peripheral T cell counts and a naive CD8 T cell phenotype and inversely with burden of regulatory and exhausted (PD-1-, CD38-, and TIM-3-expressing) T cells.50 These findings are consistent with preclinical predictions56 and with associations seen in other TCE therapies, including BCMA CAR T.57–59 The depletion of naïve and early–memory T cells, along with the increasing frequency of an exhausted immune phenotype, are common features of advanced MM and of heavily pretreated patients. The intrinsic reliance on host immunity raises the question of whether TCE therapies should be used in early lines of MM therapy rather than in the relapsed refractory setting, where their use is currently approved.
Teclistamab responders also had lower sBCMA concentrations, and elevated sBCMA (but not surface BCMA) was associated with worse disease (high-risk International Staging System scores and extramedullary involvement) and with a greater bone marrow PC cellularity.52 These features were associated with more baseline T cell dysfunction, and likely account for the lower response rates in patients with high tumour burden. Again, these findings challenge the current treatment paradigm and suggest the use of teclistamab in maintenance or other low-disease-burden setting, as investigated in the MajesTEC-4, MASTER-2 and Immuno-PRISM trials, may allow for long-lasting anti-tumour immunity (Table 1).1,51,60–71
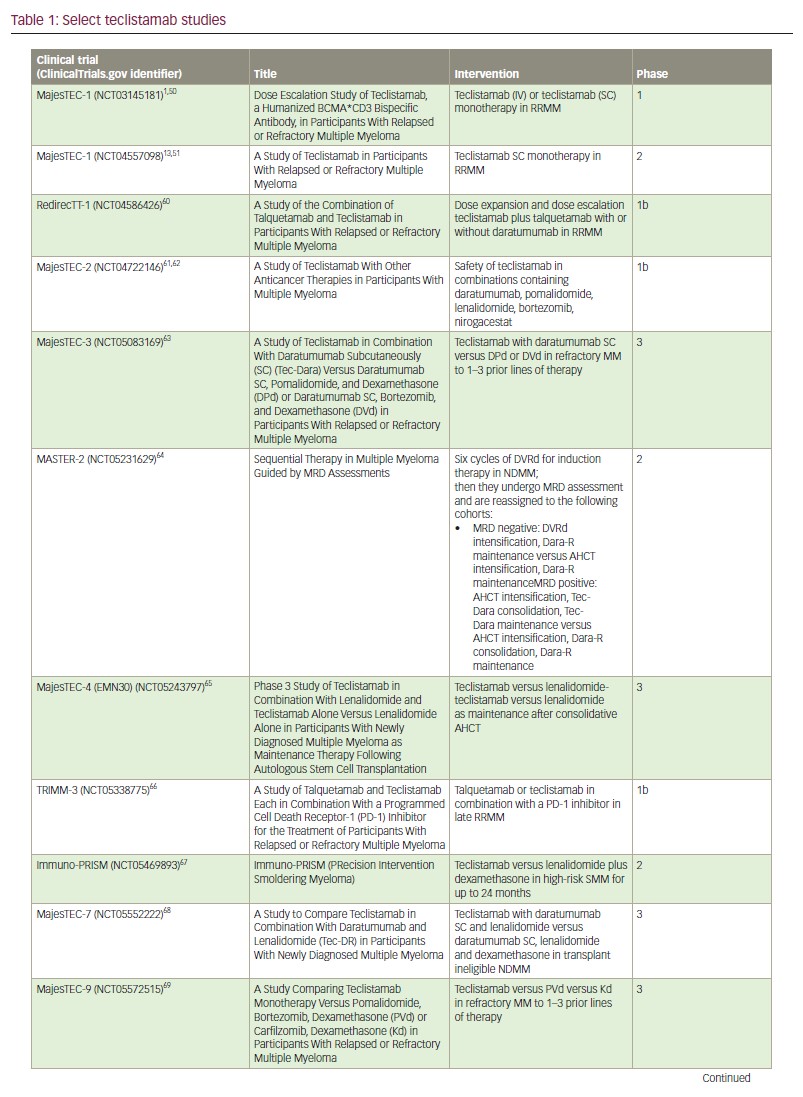
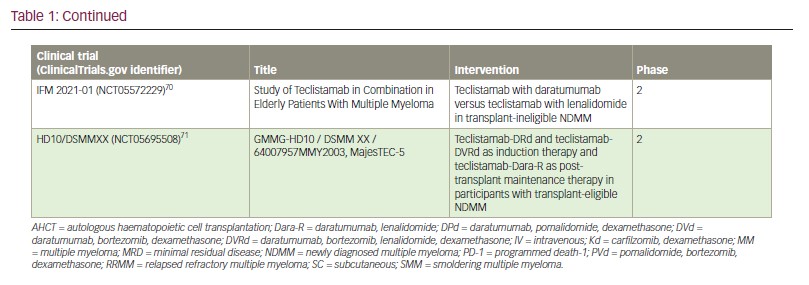
Lastly, a preclinical study found that pretreatment with CD38 inhibition enhanced teclistamab activity in a synergistic manner, possibly due to the immunomodulatory effect of daratumumab in the tumour microenvironment,72–76 and its inhibition of nicotinamide adenine dinucleotidase activity, which, in turn, may avert T cell exhaustion.40,77 Daratumumab-lenalidomide synergism has been previously demonstrated.78 As such, various teclistamab-containing multi–drug regimens are currently under investigation in newly diagnosed (MajesTEC-4,65 MajesTEC-7 [ClinicalTrials.gov identifier: NCT0555222268] and MASTER-2 [ClinicalTrials.gov identifier: NCT05231629]64), early (MajesTEC-3 [ClinicalTrials.gov identifier: NCT05083169]63 and MajesTEC-9 [ClinicalTrials.gov identifier: NCT05572515]69) and late relapsed settings (TRIMM-3 [ClinicalTrials.gov identifier: NCT05338775]66 and RedirecTT-1 [ClinicalTrials.gov identifier: NCT04586426]60) (Table 1). How and when these combinations are adopted will largely depend on the safety of the regimen and whether the progression-free and overall survival advantages outweigh the risk of incremental toxicity. For example, recently presented early results from MajesTEC-2 combining teclistamab with lenalidomide and daratumumab in patients with RRMM who had received 1–3 prior lines of therapy was highly efficacious but again associated with a high rate of infectious complications.61 In these early lines of therapy, responses are expected to be very durable, which would expose patients to cumulative immune suppression. Alternative dosing strategies in which teclistamab is administered for fixed durations, with intermittent re-dosing upon disease progression, may be important for safe use in early lines of therapy. Such intermittent dosing may also improve efficacy by alleviating T cell exhaustion associated with continuous administration.79
Treatment paradigms and discussion
TCE therapies for MM have evolved rapidly. Though CAR T cells demonstrated the most impressive single-agent response rates ever reported in RRMM just a couple of years ago, teclistamab now enables similar efficacy but with off-the-shelf availability and subcutaneous administration.9,11 Both CAR T cells and BsAb therapy continue to innovate. For example, abbreviated manufacturing protocols may improve both the efficacy and accessibility of CAR T cell therapy,80 and trispecific antibodies that enable dual-antigen specificity or that incorporate costimulatory or checkpoint-blocking domains are in development.81 Collectively, these are paradigm-shifting advances with the potential to add years to the survival of the typical patient with MM.
We do not yet know how best to use these agents in practice. Both CAR T cells and teclistamab are currently approved for patients with at least four prior lines of MM therapy; however, all these agents are being evaluated in earlier lines of MM therapy (Table 1).8,10,12 Meanwhile, non-TCE therapies continue to progress, notably with molecules such as iberdomide82 and mezigdomide,83 which build on lenalidomide’s mechanism of action, and novel immunotherapies such as modakafusp alfa, an anti-CD38 antibody-cytokine fusion protein.84 Extensive clinical investigation will be required to determine how to best sequence and combine these agents with current therapies; such studies will hopefully incorporate correlative studies to inform personalized treatment approaches.
Though it is tempting to contrast different TCE therapies, patients can receive both BsAb and CAR T cells sequentially. Teclistamab has been prospectively evaluated in a small cohort of patients previously treated with anti-BCMA CAR T cells or the anti-BCMA antibody–drug conjugate belantamab mafadotin; the response rate in these patients was only slightly lower than in patients who were naive to BCMA-directed therapy.85 Similarly, ciltacabtagene autoleucel has been prospectively studied after prior BCMA-directed therapy with promising results,86 and several retrospective reports have demonstrated efficacy with sequential BCMA-directed therapies.87–89
Although BCMA-negative relapses have been reported,90 most cases of progression after anti-BCMA CAR T cells do not appear related to antigenic escape.91 Continuously dosed BsAbs likely exert more durable target-directed immune surveillance than CAR T cells, which wane after several months in most RRMM patients.9,11 Target-negative relapse may, therefore, be more likely after BsAb therapy than CAR T cell therapy; however, this has not yet been evaluated. Patients progressing after BCMA-directed TCE therapy have been shown to respond to both BsAb and CAR T cells directed against GPRC5D.43,92,93
Our current approach to sequencing teclistamab and CAR T cells is driven by patient-specific factors, including patient preference either for a single–dose, but more complex, CAR T cell approach or for the simpler, but continuously dosed, teclistamab. Patients with rapidly progressive RRMM now have teclistamab as a readily available option. However, teclistamab may still be preferred for patients with gradual and uncomplicated progression, depending on the accessibility of a cellular therapy centre and patient tolerance of CAR T cell toxicities and logistical requirements.
Finally, with all this exciting progress, we must not lose sight of the stubbornly high propensity for relapse that remains even after treatment with these potent novel therapies and of the high cost required to sustain years of sophisticated therapy in patients with MM. More focused research is required on the specific mechanisms of relapse and the biology of resistant disease that persists, often below detectable limits, and that presumably seeds future relapses. In addition, a nuanced clinical and translational investigation is required to understand when and for whom continuous therapy promotes long-term survival compared with more intermittent therapy.