
Allogeneic stem cell transplantation (alloSCT) has been utilised in the management of leukaemia for several decades and has established its role in producing long-term remission. In the context of multiple myeloma (MM), alloSCT induces the highest rate of remissions, including molecular remission, resulting in long-term disease-free survival (DFS) in <30% of patients.1–3 However, it is associated with the highest rate of treatment-related mortality (TRM) of all of the interventions for MM, resulting from conditioning-related end-organ damage and the unwanted immunological toxicity of graft-versus-host disease (GvHD), in addition to immune-suppression-related opportunistic infection.4
The effectiveness of alloSCT results from the combination of high-dose chemotherapy and the adoptive transfer of immune-competent donor immune-effector cells (graft-versus-myeloma [GvM] effect). The former aims to cytoreduce and is combined with autologous stem-cell rescue as first-line therapy in suitably fit patients;5,6 however, all patients ultimately relapse. The GvM immunological effect is best demonstrated by the use of donor lymphocyte infusions (DLIs) to re-induce disease responses following relapse post-alloSCT. Although less potent than that seen with chronic myeloid leukaemia, it significantly contributes to the DFS effect noted with alloSCT in MM.7,8 The published evidence surrounding these two main factors in alloSCT for MM suggests that they are not mutually exclusive in their contribution to long-term DFS in MM, although the relevant contribution of each to this effect remains to be clarified. There is evidence that both the method and degree of cytoreduction and the GvM effect contribute to long-term DFS in MM. As such, the challenge for transplant physicians is how to harness the GvM effect while maintaining acceptable toxicity and extending the spectrum of patients with MM who would and could benefit from alloSCT.
The survival of patients with MM ranges from a few weeks to >20 years, but there is good evidence that the median survival of patients has increased over the last decade9 to between three and four years. This improvement is probably due to improvements in supportive care, the introduction of novel therapies and the increased use of high-dose therapy, which has made decision-making for younger patients with a human leukocyte antigen (HLA)-matched sibling donor even more difficult. MM has a median age at presentation of approximately 70 years and only 15% of patients are <60 years of age, with the result that only a minority of patients are eligible for consideration for alloSCT, even when reduced-intensity conditioning (RIC) alloSCT is included. Myeloablative alloSCT is associated with a high TRM, most likely due to a combination of factors, including advanced age of patients, myeloma-related organ damage (particularly renal impairment), disease-associated immune dysfunction and infection risk with the effects of previous treatment. Analysis of prognostic factors is essential to compare outcomes within and between clinical trials and, in particular, to assess whether certain treatments, such as alloSCT or ‘new agents’, can overcome the effect of prognostic factors that would be associated with a poor outcome with conventional therapy. The International Staging System (ISS) is considered the standard prognostic model and identifies three risk categories based on levels of β2-microglobulin and albumin.10 Cytogenetic and molecular genetic abnormalities have also been shown to be useful predictors of outcome, with the presence of chromosome 13 deletion – t(4;14), t(14;16), t(14;20) – and deletion17p by fluorescence in situ hybridisation (FISH) being associated with adverse outcome.11–13,14
Following the first alloSCT for MM in the early 1980s, a retrospective analysis of the European Group for Blood and Marrow Transplantation (EBMT) registry data with a median follow-up of 6.5 years reported that myeloablative alloSCT achieved an overall survival of 40% but was associated with a high TRM.15 A subsequent comparison of patient cohorts transplanted in 1983–1993 and 1994–1998 showed that TRM decreased from 46 to 30%4 due to better patient selection, particularly transplantation earlier in the course of the disease. Subsequently, there were several reports from single centres indicating that with stringent patient selection, better results could be obtained (see Table 1). Two prospective, randomised studies have included a biological randomisation to alloSCT. In the US Intergroup S9321 study, patients were randomised to autologous SCT, chemotherapy or Cy/total body irradiation (TBI) ablative alloSCT if a matched sibling was identified. The alloSCT arm was closed early as a result of high TRM (53%). However, a subsequent analysis with seven-year follow-up showed identical survival at 39% with slightly superior progression-free survival (PFS) in the alloSCT arm (22 versus 15%). There was also evidence of a plateau on the survival curve of the alloSCT arm.16 Similar results were obtained from a retrospective British Society of Blood and Marrow Transplantation (BSBMT) study of 139 patients <55 years of age, which showed superiority for melphalan/TBI over cyclophosphamide/TBI conditioning in both TRM and relapse rates.17 In an attempt to reduce TRM and the high incidence of GvHD, patients <55 years of age entered into the Haemato-Oncology Co-operative Group (HOVON) 24 MM study who had an HLA-matched sibling received a Cy/TBI T-cell depleted (TCD) alloSCT.18 Patients without a donor were randomised to receive a Cy/TBI autologous stem cell transplant (ASCT) or no further chemotherapy. Despite TCD, the TRM remained high at 34%, and there was a high relapse rate, with only three out of 53 alloSCT recipients in complete relapse (CR) at a median follow-up of 38 months. The results of this study suggest that profound transcranial Doppler (TCD) should be avoided.
As a result of the experience of full-intensity alloSCT, investigators explored the use of RIC alloSCT in MM to reduce TRM and to permit the application of alloSCT to older patients. Several studies have shown that this approach is feasible, with a significant reduction in TRM (see Table 1). By definition, in the absence of intensive chemo-radiotherapy, RIC alloSCT is to a large extent dependent on allogeneic GvM effects.19 A number of phase II studies have reported similar findings.20–22 In these studies, the presence of chronic GvHD was associated with the achievement of CR and improved overall survival (OS)/PFS.
Two prospective ‘biologically randomised’ studies of RIC alloSCT have been published. IFM-99-03/04 enrolled patients with poor-risk disease as defined by the presence of del13 by FISH along with elevated β2-microglobulin (>3mg/l).23 Those without a sibling donor received tandem ASCT, whereas those with a sibling donor received a sequential ASCT/RIC alloSCT, the latter conditioning utilising antithymocyte globulin (ATG) (Genzyme). This study showed no benefit in terms of OS/event-free survival (EFS) and no patients in either arm achieved durable EFS. Bruno et al. reported on a group of unselected patients who were biologically randomised in a similar fashion to tandem ASCT or a sequential ASCT/RIC alloSCT.24 The RIC alloSCT arm demonstrated significantly superior OS/EFS. While no prospective trials have compared full-intensity with RIC alloSCT in this setting, an EBMT analysis has shown similar OS with both approaches. As might be expected, RIC alloSCT had a lower TRM but a higher relapse rate and lower PFS.25
Generally speaking, outcomes with matched unrelated donor (MUD) alloSCT have improved with time and in many settings have become equivalent to matched sibling transplantation. However, retrospective studies in myeloma have shown a significantly higher TRM than sibling alloSCT,26 and myeloablative MUD alloSCT is not currently recommended and should be carried out only in the context of prospective clinical trials. However, the role of RIC MUD alloSCT remains to be defined. Encouraging results have been reported for TRM of approximately 20–21% at two and three years.27,28 Clearly, further prospective trials are warranted in order to better define the role of RIC MUD alloSCT for patients with myeloma.
No prospective trials have compared functional imaging (FI) with RIC alloSCT in MM, and the type of RIC regimen and the therapy previously administered may matter. However, an EBMT analysis has shown similar OS with both approaches, although the RIC alloSCT group had a lower TRM but a higher relapse rate and thus lower PFS.25 This may reflect the heterogeneity of the patient populations, and further adaptation of RIC alloSCT may be required to maximise the antitumour effect in association with the well-established reduced TRM. In patients >40 years of age or in those deemed unfit for an FI alloSCT, RIC alloSCT has been shown to reduce TRM. By definition, as FI conditioning is not given, RIC transplants are largely dependent on the adoptive immunotherapeutic effect of GvM. As such, a ‘tandem’ sequential strategy has been developed to generate a minimal tumour burden pre-RIC alloSCT using high-dose melphalan-conditioned ASCT. A number of phase II studies have reported the feasibility and efficacy of such a strategy, especially in association with limited chronic GvHD.20,21,25,29 Bruno and colleagues reported on a biologically randomised study of sequential ASCT/RIC alloSCT compared with double ASCT, demonstrating a superior OS and EFS with the RIC alloSCT arm.24 Two further studies are yet to be reported (European Blood and Marrow Transplantation Group [EBMT] NMAM2000, where a tandem ASCT followed by a MRD RIC alloSCT is compared with double ASCT, and the US Intergroup study). Therefore, in patients deemed to have an adequate performance status/co-morbidity profile, sequential ASCT and RIC alloSCT may be a suitable therapeutic strategy to induce a minimal residual disease (MRD) state (ASCT) on which to build the adoptive immunotherapeutic effect of GvM (RIC alloSCT).
In light of the limitations in availability of a matched related (family) donor, alternative sourcing of donor stem cell grafts is routinely employed in the management of acute and chronic myeloid malignancies. The use of matched volunteer-unrelated donor (VUD) grafts utilising FI conditioning is not generally supported even in younger patients in the first treatment phase. The improvement in VUD FI alloSCT outcomes represents a future development, but currently should be considered only as part of a clinical trial protocol.26 The role of RIC MUD alloSCT remains to be defined, although recent results have reported TRM of approximately 20%.26,27 Clearly, further prospective trials are warranted in order to better define the role of RIC MUD alloSCT for patients in the first treatment phase for myeloma. Similarly, the use of umbilical cord stem cells as a source for GvM has been reported in a limited number of case reports but only in patients with advanced disease,30,31 and again should be explored only as part of a clinical study protocol.
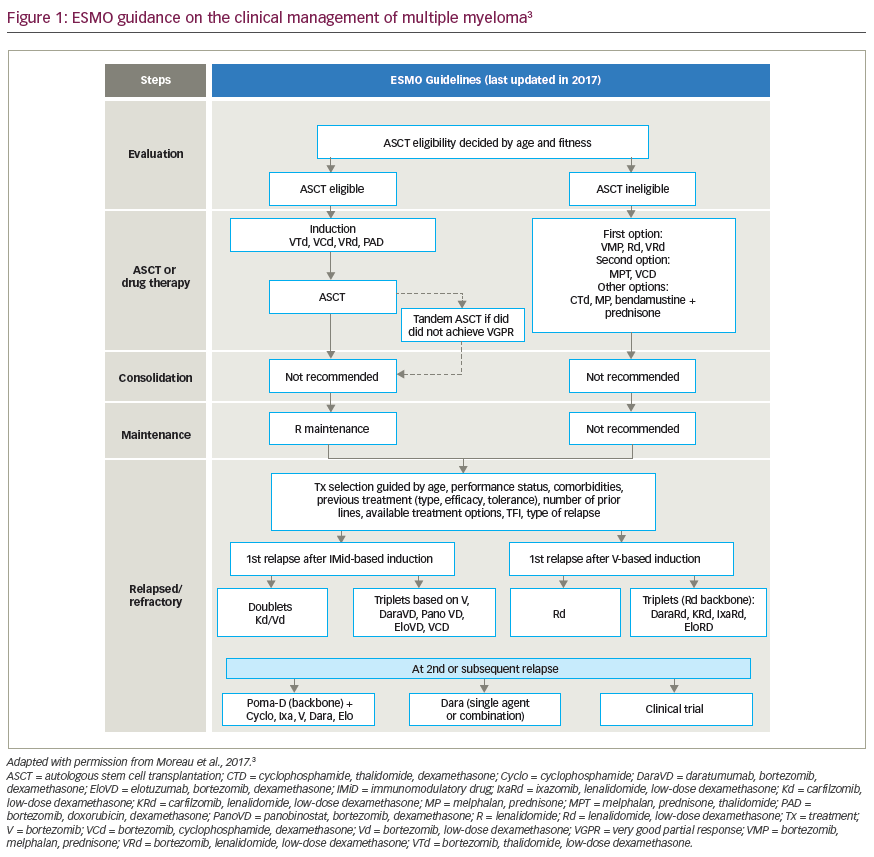
The treatment decision-making process involving the use of alloSCT in the overall management strategy of patients with MM represents some of the most difficult concerns haemato-oncology patients face. Patients need to be fully informed of the risks and potential benefits of undergoing such a treatment, and the clinical treatment strategy that is adopted should represent both the clinical evidence and the patient’s understanding and informed consent. The use of pre-transplant co-morbidity scoring systems has evolved in the stem cell transplant field, although these have yet to be validated in European populations and, specifically, their utility in a high-risk patient population such as MM remains to be determined.32,33 A potential clinical decision-making tree is represented by the flow diagram illustrated in Figure 1. Patients <40 years of age with a matched sibling donor should be considered suitable for FI alloSCT in the first treatment phase. Patients between 40 and 50 years of age may be considered suitable for FI MRD alloSCT based on biological performance status, co-morbidity and disease-related features at the discretion of the transplant physician. The optimal conditioning regimen has never been prospectively compared, but retrospective analyses favor melphalan 140mg/m2 with fractionated TBI (12Gy in six fractions).17
Is alloSCT the final therapeutic intervention for MM? Experience suggests that this in fact is more of a therapeutic platform on which to build a strategy for long-term DFS. Donor lymphocyte infusions from the original donor of the allograft are given for three main reasons: for relapse (which may be at various levels), pre-emptively in patients at high risk of relapse and for correction of mixed chimerism in order to preserve a GvM effect and reduce the chance of relapse. There has been little work aimed at defining the main effector cells (T cells or natural killer [NK] cells), which may in fact vary from disease to disease. Little is known about the main ‘antigenic’ targets and in MM many immunotherapy trials have focused on the MM-specific idiotypic determinant of the immunoglobulin (Ig) variable region, although other antigenic determinants have been suggested, such as the cancer germline antigens.34,35 The available evidence suggests that the GvM effect is ‘moderate’ and that high doses of DLI resulting in clinical acute and chronic GvHD are required to achieve sustained disease responses. Disease responses occur in up to half of relapsed patients and up to 20% achieve CR, although the median PFS can be short.7,36 Zeiser and colleagues reviewed the published myeloma donor lymphocyte infusion (DLI) data in 2004.37 Overall, 40–52% responded, with 25% having a CR. Response durations ranged from six to 15 months and responses were mainly seen after multiple DLI with high T-cell doses. Acute and chronic GvHD were seen in half and one-third, respectively. Attempts to separate the GvM and GvHD effects have been made using CD8-depleted DLI.38 Only one of nine patients who received 3×107 CD3+ cells/kg DLI had GvHD. Disease response data in patients with MM using this approach are lacking, and there will be concern that reducing GvHD will also result in fewer patients responding.
There are few reports about the use of DLI to prevent relapse (in the absence of mixed chimerism) and also few data about its use for eliminating low-level MRD detected by molecular methods. Prophylactic DLI would be expected to have substantial toxicity; however, in the setting of RIC alloSCT, the use of DLI is quite different. When these regimens involve in vivo T-cell depletion using alemtuzamab or ATG, mixed chimerism is not an uncommon outcome, and may be associated with an increased risk for relapse. The University College London group reported 109 DLIs in 46 patients (including 19 MM patients) and a chimeric response in 30 of 35 patients.39 GvHD was observed in 12 patients. Disease responses occurred in nearly two-thirds of MM patients, although surprisingly they were not necessarily associated with chimeric responses. However, much investigation is required before we can confidently advise about the dose and timing of DLI, and each disease is likely to be different.40,41 Kröger expresses the view that we should be targeting molecular remission and not settling for less rigorous assessments of disease.42 The shortcomings of these data have been noted (heterogeneity and short follow-up), and disease-specific clinical trials are required.
With the expansion of available biological therapies in the management of MM,43 the rationale for using such therapies post-alloSCT has been proposed, either pre-emptively or to treat relapsing disease.42 There are limited data concerning the use of bortezomib in this setting. In a small study, two cycles of four doses were completed by 14 patients,44 with neurotoxicity and thrombocytopenia being the most frequent reported adverse events, and in four patients there may have been some stimulation of GvHD. Nonetheless, complete and partial response rates of 30 and 50% were promising outcomes. An Italian series of 23 patients reported similar results, with 61% response and 22% immunofixation-negative CRs with PFS of six months, although similar toxicities were observed in nearly one half.45 Sixty per cent of these patients also received steroids, but this did not affect response.
There are small series reporting the use of thalidomide post-alloSCT. The InterGroupe Francophone du Myélome (IFM) group reported a 29% response rate with acceptable toxicity.46 Kröger and colleagues reported a 67% response rate with a 84% two-year PFS.47 Thalidomide has been used in several studies as a maintenance strategy post-alloSCT.48–50 Developed as an analogue of thalidomide with immunomodulatory effects, lenalidomide has been shown to be efficacious in the management of MM.51,52 However, there are fewer data concerning the use of lenalidomide in this situation. Studies are ongoing to determine the effect of post-alloSCT administration of lenalidomide either as pre-emptive therapy or for early relapsing disease, and the efficacy of this strategy will be as important as the drug-related adverse events, especially the effect, if any, on GvHD.
Although much has been learned from experience to date in the use of alloSCT in the long-term management of MM, its role is less certain than in other haematological malignancies. The overarching premises of the putative GvM effect still remain elusive without considerable risk to the recipient. Nonetheless, with all other therapy MM is a universally fatal malignancy, and while improvements have been made in OS with the introduction of new drugs, median survival is still unacceptably short. With the introduction of novel agents, the potential to take patients to alloSCT with considerably reduced tumour burdens represents the way forward, while permitting a more intensive conditioning regimen (Mini, Midi, Maxi)42 resulting from reduced pre-transplant end-organ therapy-related damage. Furthermore, the alloSCT should be viewed as the platform from which to launch a pre-emptive consolidative strategy to minimise the risk for disease relapse. This remains the challenge of the imminent future for the latter-day pioneers of alloSCT medical practice. ■
Capturing Alloreactivity in Multiple Myeloma
Abstract
Overview
Allogeneic stem cell transplantation (alloSCT) has been utilised in the management of both malignant and non-malignant haematological disorders for several decades and has established its role in producing long-term remissions. In the context of multiple myeloma (MM), compared with conventional therapies alloSCT induces the highest rate of remissions, resulting in long-term disease-free survival in over 30% of patients. However, it is associated with the highest rate of treatment-related mortality of all the interventions for MM. Following the introduction of new biological agents for the management of MM, the question of what role alloSCT has in MM is raised. This article aims to review where we are with alloSCT in MM, drawing from our experience thus far to plan the future role of alloSCT if we are to capitalise on a potential donor antimyeloma immune therapeutic effect.
Keywords
Allogeneic stem cell transplantation, multiple myeloma, adoptive immunotherapy, immunomodulatory agents
Article
References
- Corradini P, Voena C, Tarella C, et al., J Clin Oncol, 1999;17(1):208–15.
- Bensinger WI, Clin Adv Hematol Oncol, 2004;2(1):46–52.
- Rabitsch W, Prinz E, Ackermann J, et al., Eur J Haematol, 2004;72(1):26–31.
- Gahrton G, Svensson H, Cavo M, et al., Br J Haematol, 2001;113(1):209–16.
- Attal M, Harousseau JL, Facon T, et al., N Engl J Med, 2003;349(26):2495–2502.
- Child JA, Morgan GJ, Davies FE, et al., N Engl J Med, 2003;348(19):1875–83.
- Lokhorst HM, Wu K, Verdonck LF, et al., Blood, 2004;103(11):4362–4.
- Salama M, Nevill T, Marcellus D, et al., Bone Marrow Transplant, 2000;26(11):1179–84.
- Kumar SK, Rajkumar SV, Dispenzieri A, et al., Blood, 2008;111(5):2516–20.
- Greipp PR, San Miguel J, Durie BG, et al., J Clin Oncol, 2005;23(15):3412–20.
- Chiecchio L, Protheroe RK, Ibrahim AH, et al., Leukemia, 2006;20(9):1610–17.
- Gutierrez NC, Castellanos MV, Martin ML, et al., Leukemia, 2007;21(1):143–50.
- Avet-Loiseau H, Attal M, Moreau P, et al., Blood, 2007;109(8):3489–95.
- Shaughnessy JD Jr, Zhan F, Burington BE, et al., Blood, 2007;109(6):2276–84.
- Gahrton G, Tura S, Ljungman P, et al., Bone Marrow Transplant, 1991;7(Suppl. 2):32.
- Barlogie B, Zangari M, Bolejack V, et al., Clin Lymphoma Myeloma, 2006;6(6):469–74.
- Hunter HM, Peggs K, Powles R, et al., Br J Haematol, 2005;128(4):496–502.
- Lokhorst HM, Segeren CM, Verdonck LF, et al., J Clin Oncol, 2003;21(9):1728–33.
- Maloney DG, Molina AJ, Sahebi F, et al., Blood, 2003;102(9):3447–54.
- Mohty M, Boiron JM, Damaj G, et al., Bone Marrow Transplant, 2004;34(1):77–84.
- Perez-Simon JA, Caballero D, Mateos MV, San Miguel JF, Leuk Lymphoma, 2004;45(9):1725–9.
- Gerull S, Goerner M, Benner A, et al., Bone Marrow Transplant, 2005; 36(11):963–9.
- Frederic Garban MA, Michallet M, Hulin C, et al., Blood, 2006;107(9):3474–80.
- Bruno B, Rotta M, Patriarca F, et al., N Engl J Med, 2007;356(11):1110–20.
- Crawley C, Iacobelli S, Bjorkstrand B, et al., Blood, 2007;109(8):3588–94.
- Shaw BE, Peggs K, Bird JM, et al., Br J Haematol, 2003;123(5):886–95.
- Kroger N, Sayer HG, Schwerdtfeger R, et al., Blood, 2002;100(12):3919–24.
- Bruno B, Sorasio R, Patriarca F, et al., Eur J Haematol, 2007;78(4):330–37.
- Alyea E, Weller E, Schlossman R, et al., Bone Marrow Transplant, 2003;32(12):1145–51.
- Fenk R, Neumann F, Fenk B, et al., Leuk Res, 2008;32(7):1157–9.
- Ando T, Yujiri T, Tominaga T, et al., Eur J Haematol, 2005;74(2):175–9.
- Sorror ML, Maris MB, Storb R, et al., Blood, 2005;106(8):2912–19.
- Parimon T, Au DH, Martin PJ, Chien JW, Ann Intern Med, 2006;144(6):407–14.
- Goodyear O, Piper K, Khan N, et al., Blood, 2005;106(13): 4217–24.
- Atanackovic D, Arfsten J, Cao Y, et al., Blood, 2007;109(3): 1103–12.
- Kuruvilla J, Shepherd JD, Sutherland HJ, et al., Biol Blood Marrow Transplant, 2007;13(8):925–31.
- Zeiser R, Bertz H, Spyridonidis A, et al., Bone Marrow Transplant, 2004;34(11):923–8.
- Alyea EP, Canning C, Neuberg D, et al., Bone Marrow Transplant, 2004;34(2):123–8.
- Peggs KS, Thomson K, Hart DP, et al., Blood, 2004;103(4):1548–56.
- Peggs K, Mackinnon S, Leukemia, 2004;18(9):1541–2, author reply 1542–3.
- Marks DI, Lush R, Cavenagh J, et al., Blood, 2002;100(9): 3108–14.
- Kroger N, Mini-Midi-Maxi? Leukemia, 2007;21(9):1851–8.
- Mitsiades CS, Hayden PJ, Anderson KC, Richardson PG, Best Pract Res Clin Haematol, 2007;20(4):797–816.
- van de Donk NW, Kroger N, Hegenbart U, et al., Bone Marrow Transplant, 2006;37(12):1135–41.
- Bruno B, Patriarca F, Sorasio R, et al., Haematologica, 2006;91(6):837–9.
- Mohty M, Attal M, Marit G, et al., Bone Marrow Transplant, 2005;35(2):165–9.
- Kroger N, Shimoni A, Zagrivnaja M, et al., Blood, 2004;104(10):3361–3.
- Attal M, Harousseau JL, Leyvraz S, et al., Blood, 2006;108(10):3289–94.
- Brinker BT,Waller EK, Leong T, et al., Cancer, 2006;106(10):2171–80.
- Feyler S, Rawstron A, Jackson G, et al., Br J Haematol, 2007;139(3):429–33.
- Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, et al., Br J Haematol, 2008;140(1):36–45.
- Weber DM, Chen C, Niesvizky R, et al., N Engl J Med, 2007;357(21):2133–42.
- Kroger N, Einsele H, Wolff D, et al., Bone Marrow Transplant, 2003; 31(11):973–9.
- Kroger N, Schwerdtfeger R, Kiehl M, et al., Blood, 2002;100(3):755–60.
- Lee CK, Badros A, Barlogie B, et al., Exp Hematol, 2003;31(1):73–80.
Article Information
Disclosure
The author has no conficts of interest to declare.
Correspondence
Gordon Cook, MBChB, PhD, FRCP, FRCPath, FRCPI, Director, Blood and Marrow Transplantation, St James’s Institute of Oncology, Bexley Wing, St James’s University Hospital, Leeds, LS9 7TF, UK. E: Gordon.Cook@leedsth.nhs.uk
Received
2009-03-27T00:00:00
Further Resources

Trending Topic
Multiple myeloma (MM) is the second most common haematological malignancy, with upwards of 35,000 diagnoses in the USA each year.1,2 It remains a leading cause of blood cancer–related mortality worldwide, and although therapeutic advances have allowed for significant improvements in the median overall survival,3,4 the majority of patients still experience cycles of relapse that are eventually fatal.5 While patients with MM are living longer, a subgroup with high-risk disease at diagnosis still does poorly, with a median […]

The optimal maintenance approach postautologous stem cell transplantation (ASCT) for patients with multiple myeloma is still an area of ongoing research. Lenalidomide has demonstrated clear superiority in clinical trials compared to placebo, and continuous post-transplant lenalidomide until progression is considered ...
Treatment options for myeloma have rapidly expanded in the past decade and, as with many cancers, the number of oral drug options has increased considerably. Given the more limited number of cancer hospitals, patients often must travel considerable distance to ...
Multiple myeloma (MM) remains largely an incurable disease with only a small percentage of patients achieving long-term remission.1 Here, we highlight some of the major studies on MM presented at the American Society of Hematology meeting in December 2021. Monoclonal gammopathy ...

Background on anti-BCMA CAR-T therapy Despite notable advances in the development of new drugs and the improvement of survival rates over the last 20 years, multiple myeloma (MM) persists as an incurable disease and nearly 35,000 new cases are expected in the ...
Multiple myeloma (MM) is the second most common haematological malignancy, affecting an estimated 450,600 patients worldwide, with an estimated 34,920 new diagnoses and 12,410 patient deaths in the USA in 2021.1,2 Although established curative therapies for MM have not yet been defined, we have ...
Multiple myeloma is a plasma cell malignancy that typically develops in individuals in their late 60s with an average survival time of ~8 years.1 Despite recent advances in treatment, multiple myeloma remains largely incurable due to development of drug resistance in ...
Multiple myeloma (MM) is a malignant clonal disorder of plasma cells in the bone marrow, and more than 32,000 new cases are expected in the USA each year.1 Many clinical advances, including novel drugs and continuous therapy with treatment sequencing, have ...

Multiple myeloma (MM) is the second most common hematological malignancy after non-Hodgkin’s lymphoma.1 Although recent decades have seen considerable advances and improvements in clinical outcomes for patients with MM,2 MM remains incurable, with a high disease burden.3 Clonal evolution ...
Amyloidosis is a group of diseases resulting from intracellular and extracellular deposition of insoluble abnormal amyloid fibrils, which alters the normal function of tissues. Glycosaminoglycans, apolipoprotein-E and serum amyloid P component comprise 10% of deposits, while amyloid fibrils formed by misfolded ...
An ever-expanding understanding of the biological basis for multiple myeloma (MM) combined with a widening array of effective therapies has dramatically improved outcomes for patients and has provided new opportunities for disease segmentation and targeted therapeutics.1 While this progress is ...
Smoldering myeloma is an asymptomatic clonal plasma cell disorder that is characterized by the presence of ≥3 g/dl serum M-protein and/or 10–59% bone marrow plasma cell infiltration, and is a precursor stage to multiple myeloma.1 Historically, patients with smoldering myeloma ...
The past decade has experienced a shift in the treatment landscape for multiple myeloma (MM), with the introduction of novel agents and new combination therapies which have improved survival outcomes for patients across all disease settings.1 These advances in treatment ...
Log into your Touch Account
Earn and track your CME credits on the go, save articles for later, and follow the latest congress coverage.
Sign up with an Email
Or use a .
This Functionality is for
Members Only
Explore the latest in medical education and stay current in your field. Create a free account to track your learning.
