Epigenetics encompasses heritable changes in the pattern of gene expression mediated by mechanisms other than alterations in primary nucleotide sequence. The epigenome is an inheritable record of changes to the DNA and histone proteins, such as methylation and nucleosome remodelling that directs which genes are to be silenced or expressed. Following early studies of abnormal gene expression in cancer, epigenetic modifications have been recognised to be of central importance in cancer pathophysiology.1 Mutations in regulators of the epigenome have been identified in cancer that have roles such as ‘writers’, ‘readers’, ‘erasers’ or ‘editors’ and have the potential to deregulate the expression of hundreds of genes genome-wide.2
Multiple myeloma (MM), a clonal expansion of plasma cells, is characterised by monoclonal protein production, end-organ damage and marked clinical and genetic heterogeneity.3–5 MM is the second most common haematological malignancy6 and accounts for approximately 20% of deaths from haematological malignancies7 and 0.8% of deaths from all cancers;8,9 in addition, MM is associated with high costs in healthcare provision.10 The median age of MM diagnosis is 74 years.11 In Europe, between 2005 and 2009, there was an over two-fold difference between the highest male cancer mortality in Hungary (235.2/100 000) and the lowest mortality in Sweden (112.9/100 000), and a 1.7-fold one in women (from 124.4 in Denmark to 71.0/100 000 in Spain).12Over the past decade, however, survival of patients with MM has improved dramatically with the advent of newer therapies.13
MM always follows the premalignant state of monoclonal gammopathy of undetermined significance (MGUS), although the precise molecular mechanisms involved in the progression from MGUS to MM are not properly understood. Various distinct genetic abnormalities have been reported in both MM and MGUS including epigenetic alterations such as DNA and histone methylation, and are known to contribute to the pathogenesis of the disease.14–17 Front-line treatment of MM can result in high response rates,18–21 although all patients eventually relapse and more effective therapies are needed. MM is heterogeneous and is associated with complex gene abnormalities and multiple signalling aberrations. A strategy of targeting single signalling pathways, genes or individual gene products may not therefore be inadequate to supress MM cell growth.22 The aim of this review is to describe epigenetic modifications and therapeutic combination strategies in MM.
Mb>DNA methylation
DNA methylation, which is the best characterised epigenetic modification, occurs in cytosinecontaining nucleotides that are immediately followed by nucleotide sequences containing guanine (i.e., cytosine-phosphodiester bond-guanine [CpG] islands). Located in 60% of promoters, CpG islands, when methylated usually results in silencing of tumour suppressor genes.23 These events are more frequent than tumour suppressor gene mutations. The CpG island methylator phenotype (CIMP), was first identified in colorectal cancer24 and has been observed extensively in a wide variety of tumours,25 although CIMP has rarely been reported in MM.26 MM is however characterised by a level of heterogeneity in DNA methylation that exceeds that described in several solid cancers.27,28
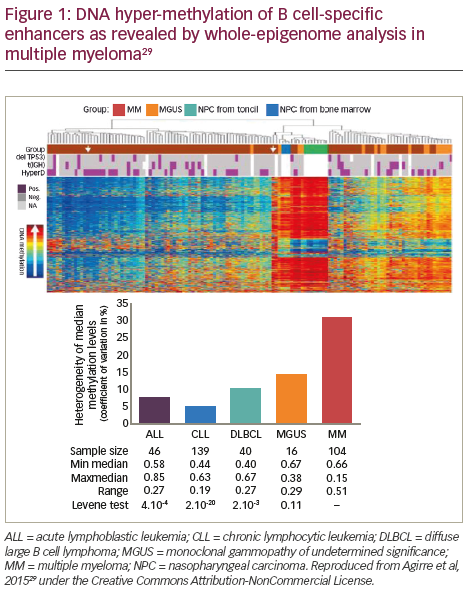
The DNA methylome, which is the map of DNA methylation modifications, has been described in normal plasma cells (NPCs) and plasma cells from MGUS and MM patient samples (Figure 1).29 Aberrant methylation patterns are related to disease stage in MM with significant difference and heterogeneity in the global methylation patterns in the malignant cell types (MM, primary plasma cell leukaemia and in human myeloma cell lines) compared to the non-malignant cells (non-plasma cell disorders and MGUS).17 DNA methylation at transcription start sites and gene promoters has been associated with the formation of heterochromatin and long-term gene silencing.30,31 Methylation is also an inactivating mechanism of the tumour suppressor p16 gene in MM, which is associated with high plasma cell proliferation and short survival.32 Furthermore, p16 methylation may be a marker for overall epigenetic changes associated with disease progression.33 Global methylation analysis in MM has pinpointed inactivated tumour suppressor genes that are prognostically important.28 In an analysis of the association of differential DNA methylation with prognosis in 159 patients with myeloma, 195 genes with changes in DNA methylation were identified. Hypermethylation of the genes GPX3, RBP1, SPARC and TGFBI was associated with significantly shorter overall survival (OS); this association was independent of age, International Staging System score, and adverse cytogenetics.28
The most comprehensive methylation study to date has demonstrated that B-cell specific intronic enhancers undergo DNA hyper-methylation, resulting in enhanced decommissioning of genes related to B-cell differentiation consistent with MM acquiring or preserving a ‘stem cell-like’ methylome.30 Finally, the expression of DNA (cytosine-5-)- methyltransferase 1 (DNMT1) is also known to be upregulated during disease pathogenesis compared to NPCs and silencing of DNMT1 significantly reduced the methylation of p16.34,35
MicroRNA are a class of short non-coding RNA molecules with RNA silencing functions.36 Epigenetic dysregulation of tumour-suppressor microRNA genes via DNA methylation has also been implicated in MM.37
Histone modifications
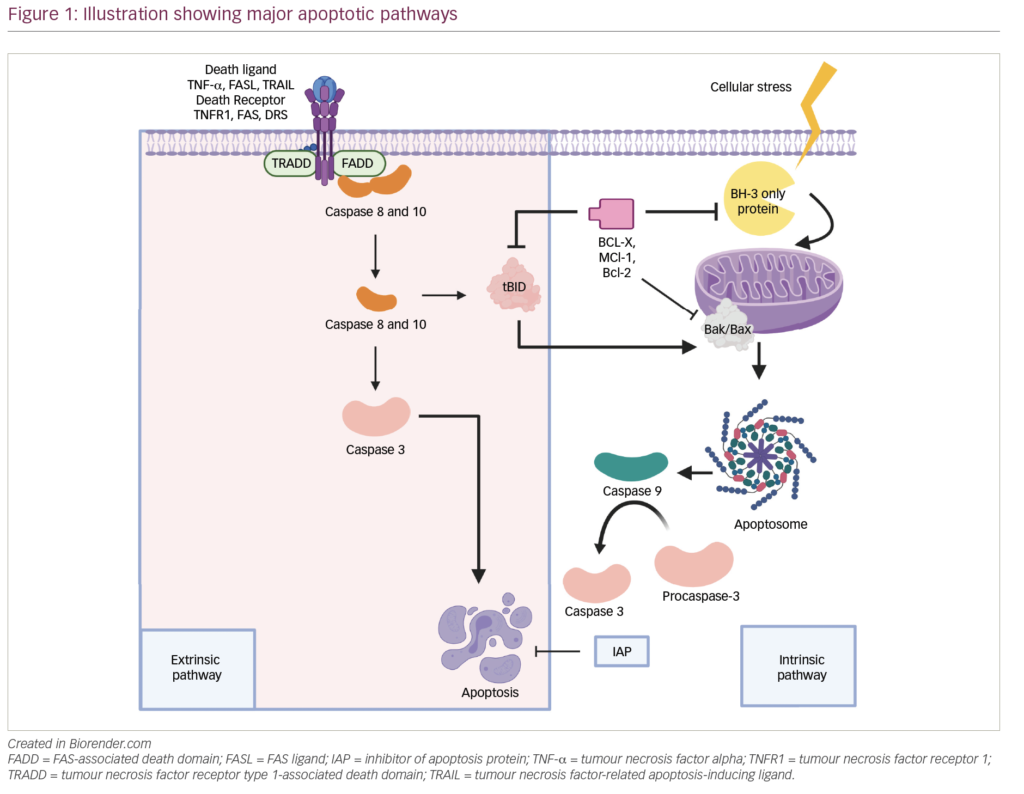
Histone modifications, including methylation, de/acetylation, phosphorylation, ubiquitination, adenosine diphosphate (ADP)-ribosylation and isomerisation, modulate gene expression by altering chromatin structure or recruiting histone modifiers.38 MMSET domain (MMSET) is a histone methyltransferase that is overexpressed in t(4;14)+ MM patients and is considered a key driving factor in the pathogenesis of this subtype of MM. MMSET catalyses dimethylation of lysine 36 on histone H3 (H3K36me2), and its overexpression causes a global increase in H3K36me2 and a decrease in lysine 27 methylation, resulting in gene expression changes promoting disease progression and tumoregenicity.39–43 Conversely, suppression of MMSET affects MM cell survival through regulation of genes associated with cell cycle progression, apopotosis and cell adhesion.44
Histone deacetylases (HDAC) are a highly conserved group of enzymes, currently grouped into four classes, that influence a wide variety of cellular functions via their ability to acetylate both histones and cytoplasmic proteins.45 These functions include proliferation, differentiation and apoptosis. Moreover, overexpression of Class I HDAC expression has
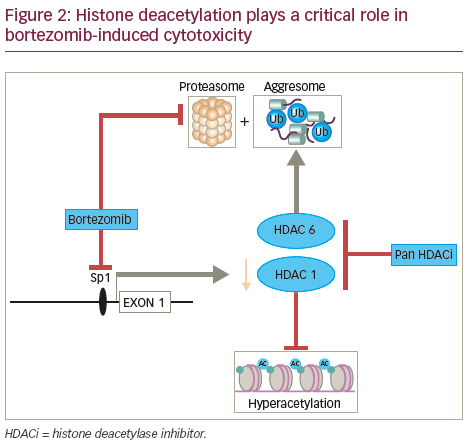
been associated, in most but not all cases, with locally advanced, dedifferentiated, proliferative tumours46 and with poor prognosis in MM.47 In contrast, expression of Class II HDACs has been found to be reduced in tumours and high expression of these isoforms has been linked in some instances to improved patient outcome in a range of both solid and haematological malignancies highlighting the contextual impact of HDAC dysregulation in cancer.46 In addition, the Class I specific HDAC inhibitor romidepsin has been shown to induce histone acetylation by promoting Caspase 8-mediated destruction of a critical Class I HDAC transcription factor Sp1.48 The combination of bortezomib and romidepsin thus promoting histone hyper-acetylation coincident with the putative disruption of protein homeostasis that such a combination would also induce (see Figure 2).
Therapeutic strategies in multiple myeloma
DNA and histone methylation
During cell division, DNMT1 maintains the DNA methylation pattern of the newly synthesised daughter strand, whereas the histone–lysine methyltransferase G9a (EHMT2) methylates histone H3 Lys9, with direct interaction between DNMT1 and G9a known to coordinate DNA- and histone-methylation during replication.49 Dual and reversible inhibitors are thus being developed to target G9a and DNMT1. Currently available DNMT inhibitors, 5-azacytidine and 5-aza-2’deoxycytidine have been evaluated pre-clinically in both in vitro and in vivo studies against MM and the DNA methylation score was shown to be predictive of MM cell sensitivity to 5-azacitidine.50,51 Oral 5-azacitidine in combination with lenalidomide and dexamethasone has been demonstrated to be effective and well tolerated in a group of heavily pre-treated patients with relapsed and/or refractory MM (RRMM) refractory to prior lenalidomide therapy.52 The Histone methyltransferase MMSET is known to be upregulated in t(4;14) patients and silencing of MMSET results in anti-MM activity,41,44,53 and while MMSET is currently not targetable, histone-methylation modifying drugs are under development.
HDAC inhibition
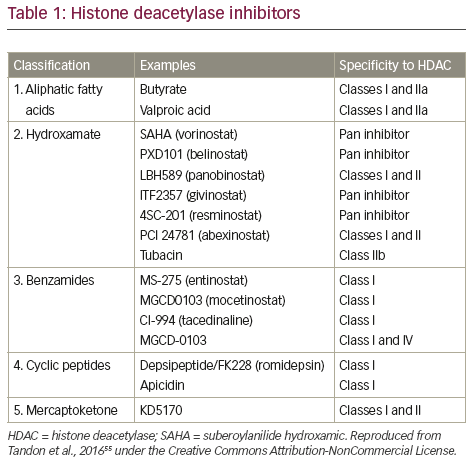
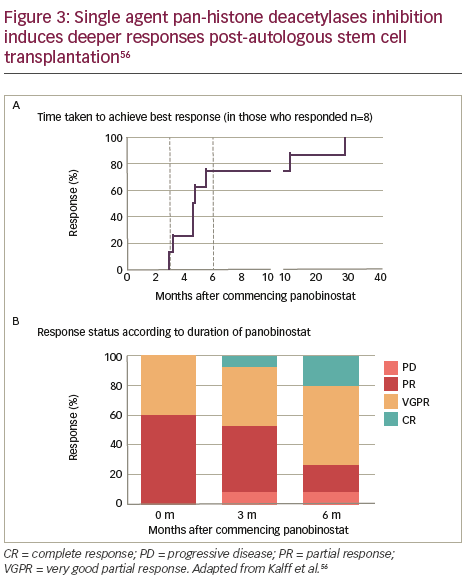
In vitro evaluation of a range of HDAC inhibitors (HDACi) has demonstrated that Class I HDAC inhibition is required by HDACi to induce maximal MM cell death.54 Major HDACi can be divided in to five categories on the basis of their chemical structure (Table 1). 55 The single-agent pan-HDACi, panobinostat (LBH589), in an interim analysis of a Phase II study was shown to induce deeper responses following high-dose chemotherapy conditioned autologous stem cell transplantation as demonstrated by both the time taken to achieve best response (Figure 3A) and response status according to duration of treatment with panobinostat (Figure 3B).56 Panobinostat has recently been approved by the US Food and Drugs Association, and European Union marketing authorisation was granted on 28 August 2015 for use in patients with MM who had received ≥2 prior therapeutic regimens that included bortezomib and an immunomodulatory drug. To avoid side effects associated with broad HDAC inhibition, class- and/or isoform-selective HDACi have been developed such as ricolinostat (ACY-1215) a selective HDAC6 inhibitor, which, coupled with bortezomib, as discussed below, targets ubitquinated protein degradation in proteasomes and lysosomes
Combination strategies
Histone deacetylase inhibition and bortezomib
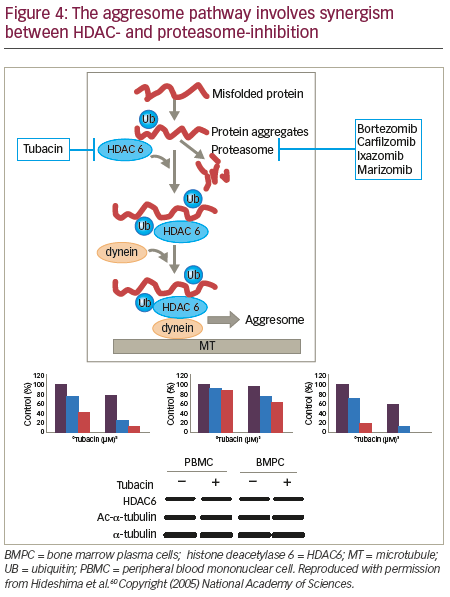
Recent studies have demonstrated an alternative system to the proteasome for degradation of polyubiquitinated misfolded and/ or unfolded proteins: the aggresome (Figure 4).57,58 HDAC6 has an essential role in a variety of cellular processes, including aggresomal protein degradation.59 Selective inhibition of HDAC6 leads to increased acetylation of its substrate, tubulin, and inhibition of the proteasome pathway, resulting in accumulation of polyubiquitinated proteins and apoptosis in MM cells.60 Inhibition of both proteasomal and aggresomal protein degradation systems by bortezomib and tubacin (a HDAC6 inhibitor), respectively, have been shown to induce synergistic cytotoxicity in MM cell lines and freshly isolated bone marrow plasma cells (BMPCs) from MM patients.60 This finding provides the rationale for clinical protocols of this combination to improve patient outcome in MM. Further, the synergistic activity of HDACi and proteasome inhibitors has been demonstrated in two mouse models that were created via subcutaneously injected human MM and intravenously injected luciferase-expressing human MM.61 Tumour growth was delayed significantly and OS was significantly prolonged in mice treated with the combination therapy. Ricolinostat (ACY-1215) is a HDAC6 inhibitor. In a phase Ib study, the ricolinostat– bortezomib–dexamethasone combination induced a treatment response in a group of heavily pre-treated patients with RRMM and had a manageable tolerability profile.62 Forty-two patients were enrolled to eight combination cohorts: the first cohort received ricolinostat 40 mg once daily (QD) with bortezomib 1.0 mg/m2; subsequent cohorts were treated with bortezomib 1.3 mg/m2 with ricolinostat doses of 40, 80, 160 and 240 mg QD, and 160 mg twice daily (BID). Ricolinostat was administered orally on days 1–5 and 8–12 of a 21- day cycle, with bortezomib on days 1, 4, 8, 11 and dexamethasone on days 1, 2, 4, 5, 8, 9, 11 and 12. Among the 25 patients evaluable for response, the overall response rate was 44%: two patients had a very good partial response, nine had a partial response, and two had a
minimal response, giving a clinical benefit rate of 52%. Diarrhoea was the only dose-related adverse event.
The HDACi, vorinostat (suberoylanilide hydroxamic acid, Zolinza®, Merck, New Jersey, US), in combination with bortezomib, showed limited clinical benefit in the multicentre, randomised, double-blind study, VANTAGE 088.63 Patients were allocated randomly to receive 21-day cycles of bortezomib (1.3 mg/m2 intravenously on days 1, 4, 8, and 11) in combination with oral vorinostat (400 mg; n=315) or matching placebo (n=320) QD on days 1–14. Median progression-free survival (PFS) was 7.63 months (95% confidence interval [CI] 6.9–8.4) for vorinostat + bortezomib and 6.83 (95% CI 5.7–7.7) for placebo + bortezomib with a hazard ratio (HR) of 0.774 (0.64–0.94) p=0.01. The most common grade 3–4 adverse events were thrombocytopenia (143 [45%] patients in the vorinostat group versus 77 [24%] patients in the placebo group), neutropenia (89 [28%] versus 80 [25%]), and anaemia (53 [17%] versus 40 [13%]). In total, 312 (99%) of 315 patients in the vorinostat group and 315 (98%) of 320 patients in the placebo group had adverse events.
VANTAGE 095, an open-label, single-arm, multicentre, phase IIb study, was designed to investigate the efficacy and tolerability of oral vorinostat with bortezomib in patients with MM that was considered refractory to novel myeloma agents.64 Patients received 21-day cycles of bortezomib (1.3 mg/m2 intravenously on days 1, 4, 8, and 11) and oral vorinostat (400 mg/d on days 1-14). The overall response rate (ORR) was 11.3% (95% confidence interval, 6.6%-17.7%), and the median duration of response was 211 days (range, 64-550 days); the median OS duration was 11.2 months (95% CI, 8.5-14.4 months), with a 2-year survival rate of 32%. Frequently reported adverse events included thrombocytopenia (69.7%), nausea (57.0%), diarrhoea (53.5%), anaemia (52.1%) and fatigue (48.6%).
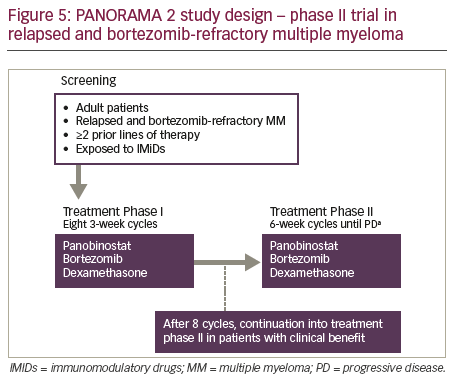
PANORAMA 2 was a phase II trial of panobinostat in combination with bortezomib and dexamethasone to treat patients (n=55) with RRMM (with ≥2 prior lines of therapy, including an immunomodulatory drug, and patients who had progressed on or within 60 days of the last bortezomibbased therapy).65 Patients were treated in two phases: Phase I treatment consisted of 8 three-week cycles of oral panobinostat (20 mg) three times per week on weeks 1 and 2, bortezomib (1.3 mg/m2 intravenously) two times per week on weeks 1 and 2, and oral dexamethasone (20 mg) four times per week on weeks 1 and 2 on days of and after bortezomib use. Patients who showed clinical benefit in phase I treatment continued study therapy in phase II, which consisted of 6-week cycles of panobinostat three times per week on weeks 1, 2, 4 and 5; bortezomib once per week on weeks 1, 2, 4 and 5; and dexamethasone on the days of and after bortezomib until disease progression, death, toxicity or treatment
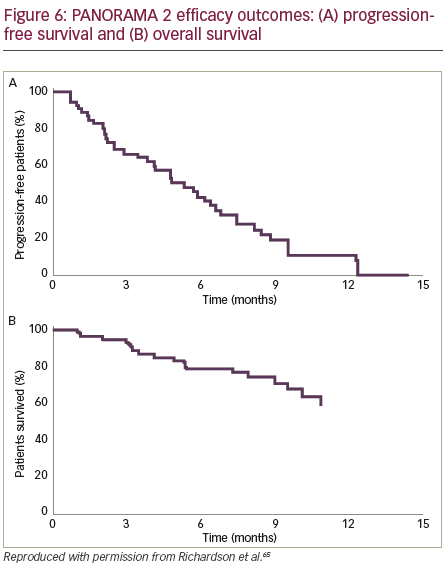
withdrawal (Figure 5). The efficacy outcomes (Figure 6) showed that panobinostat, when combined with bortezomib and dexamethasone, can recapture responses in heavily pre-treated, bortezomib-refractory MM patients.
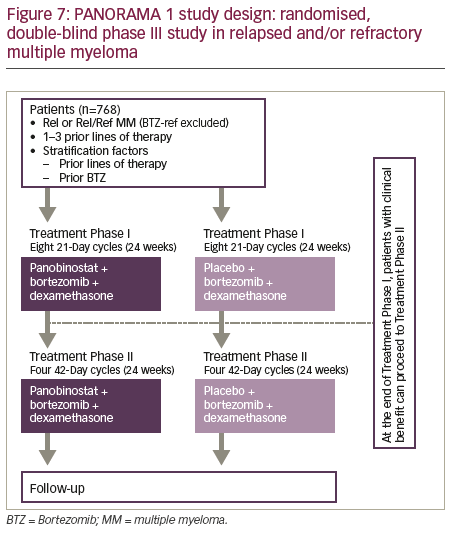
PANORAMA 1 was a multicentre, randomised, placebo-controlled, doubleblind phase III trial of patients (n=768) with RRMM who had received between one and three prior lines of therapy (Figure 7).66 Patients were randomised to receive 21-day cycles of placebo or panobinostat (20 mg; on days 1, 3, 5, 8, 10, 12, orally), both in combination with bortezomib (1.3 mg/m2 on days 1, 4, 8 and 11, intravenously) and dexamethasone (20 mg on days 1, 2, 4, 5, 8, 9, 11, 12, orally). The primary endpoint of PFS was met (p<0.0001) with a clinically relevant increase in median PFS of 3.9 months for the panobinostat–bortezomib–dexamethasone arm. Serious adverse events (SAEs) were reported in 228 (60%) of 381 patients in the panobinostat–bortezomib–dexamethasone group and 157 (42%) of 377 patients in the placebo–bortezomib–dexamethasone group. Common grade 3–4 laboratory abnormalities and adverse events (irrespective of association with study drug) included thrombocytopenia (256 [67%] in the panobinostat group versus 118 [31%] in the placebo group), lymphopenia (202 [53%] versus 150 [40%]), diarrhoea (97 [26%] versus 30 [8%]), asthenia or fatigue (91 [24%] versus 45 [12%]), and peripheral neuropathy (67 [18%] versus 55 [15%]). Nearly half (48.4%) had received ≥2 prior lines of therapy. A post-hoc subgroup analysis revealed a relatively greater increase in PFS in heavily pre-treated patients who had received ≥2 prior lines of therapy and both prior bortezomib and an immunomodulatory drug with a median improvement in PFS associated with the use of panobinostat of 7.8 months.67
HDACi and immunomodulatory drug combinations
In a Phase II trial in RRMM (n=20), the all oral combination of panobinostat– lenalidomide–dexamethasone demonstrated durable responses, even in high risk lenalidomide refractory patients, indicating the possible capacity of panobinostat to overcome acquired drug resistance.68 The treatment regimen consisted of panobinostat 20 mg on days 1, 3,
5, 15, 17, 19 together with lenalidomide 25 mg qd on days 1–21 and dexamethasone 40 mg on days 1, 8, 15 in a 28-day cycle. The primary endpoint, ORR, was 45%. Common grade 3–4 toxicities were neutropenia (55%), thrombocytopenia (40%) and infection (20%) and common grade 3–4 diarrhoea occurred in 15%. One patient discontinued due to an adverse event (asymptomatic QTc prolongation).
Ricolinostat in combination with lenalidomide and dexamethasone also demonstrated efficacy in patients (n=25) with RRMM.69 In the first phase of the study, patients were treated with escalating doses of ricolinostat on days 1–5 and 8–12 of a 28-day cycle with lenalidomide 25 mg on days 1–21 and dexamethasone 40 mg weekly. In the second phase, ricolinostat was also given on days 15–19. Doses up to 240 mg QD and 160 mg BID were explored. The ORR was 64% and 50% in the full cohort and in a lenalidomide-refractory patient cohort, respectively. Adverse events were generally low grade and manageable; common grade 3–4 toxicities included neutropenia and fatigue. Two dose-limiting toxicities have been reported to date (grade 3 syncope and grade 3 muscle cramps).
Panobinostat and multidrug combinations
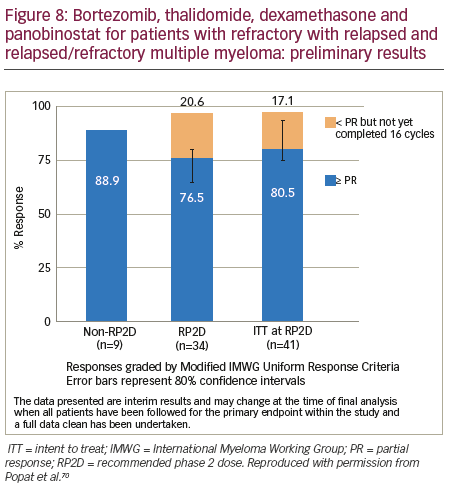
The combination of bortezomib–thalidomide–dexamethasone–panobinostat combination is being evaluated in an ongoing Phase I/II study (n=55).70 The ORR was 88.9% in the dose escalation phase and currently >75% for patients being treated at the recommended phase II dose (RP2D) (Figure 8). The regimen comprised bortezomib 1.3 mg/m2 subcutaneously days 1 and 8, thalidomide 100 mg/daily (50 mg if pre-existing neuropathy), dexamethasone 20 mg days 1, 2, 8, 9 and panobinostat days 1, 3, 5, 8, 10, 12) every 3 weeks. Grade 3–4 adverse events were primarily haematological: hypophosphatemia (16.7%), neutropenia (14.6%), abnormal liver tests (6.3%) (none observed at the non-recommended dose); thrombocytopenia (6.3%), peripheral neuropathy (2.1%), diarrhoea (2.1%) and hyperglycaemia (2.1%). The predominant grade 1–2 toxicities were: fatigue (64.5%), neuropathy (28, 58.3%), constipation (22, 45.8%) and diarrhoea (35.4%). Nineteen SAEs were reported by 12 patients at the RP2D, of which four were treatment related and predominantly gastrointestinal. The RP2D for panobinostat in combination with thalidomide, dexamethasone and bortezomib was 20 mg. Only one dose-limiting toxicity was observed; therefore, the maximum tolerated dose was not reached. The panobinostat–bortezomib–lenalidomide–dexamethasone combination has been investigated in patients (n=22) with newly diagnosed MM.71 The maximum tolerated dose without supportive care was determined to be: lenalidomide 25 mg; bortezomib 1.3 mg/m2; dexamethasone 20 mg and panobinostat 10 mg. This multidrug combination led to rapid, high-depth responses with a near complete response (CR)/CR rate of 50% and was very well tolerated with limited grade 3–4 adverse events. One patient experienced grade 4 thrombocytopenia, and a second patient had grade 3 diarrhoea without supportive measures, for < 12 hours that subsequently resolved with supportive measures. This is the first trial that has combined panobinostat with subcutaneous bortezomib, which potentially contributed to the favourable adverse event profile.
HDAC inhibitors and carfilzomib
In vitro, ricolinostat and carfilzomib combination treatment led to synergistic anti-MM effects and an in vivo mouse xenograft study confirmed a decrease in tumour volume, associated with apoptosis, providing additional support for the combination of deacetylase and proteasome inhibitors.72 The combination of panobinostat and carfilzomib was shown to be feasible and effective in patients with RRMM in a phase I/II study.73 ORR in all patients (n=35) was 77% and common grade 3–4 haematological adverse events were thrombocytopenia (47%) and neutropenia (8%). Two additional ongoing studies are evaluating the combination of panobinostat and carfilzomib (ClinicalTrials.gov Identifiers: NCT01549431 and NCT01496118) and are expected to complete late-2016.74,75
HDAC inhibitors and pomalidomide
In vitrodata have suggested synergism between the HDACi, ricolinostat, and both lenalidomide and pomalidomide.76 Two clinical trials of ricolinostat in combination with pomalidomide and dexamethasone in RRMM (planned n=20 and n=100) are ongoing (NCT 02189343 and NCT 01997840).
Epigenetic therapies in combination with monoclonal antibodies
HDACi have been shown to modulate the anti-tumour immune responses through the engagement of natural killer group 2D (NKG2D) receptors with their ligands.77,78 HDACi also upregulate antigen expression on tumour cells.79,80 It has been hypothesised that HDAC inhibition will increase antibody-dependent cell-mediated cytotoxicity by upregulating NKG2D ligand and CD38 expression on MM cells thereby contributing to improved efficacy of monoclonal antibody treatment during induction and especially during consolidation and maintenance. Finally, panobinostat has been shown to upregulate the MM-specific kappa myeloma antigen in vitro and subsequently enhance antibodydependent cellular cytotoxicity with the developmental monoclonal antibody MDX1097.81
In mice studies, both anti-PD-1 and anti-cytotoxic T-lymphocyteassociated antigen-4 (CTLA-4) antibodies were unable to eradicate large, modestly immunogenic CT26 tumours or metastatic 4T1 tumours.82 Co-treatment with epigenetic-modulating drugs (5-azactytidine, a DNA methyltransferase inhibitor) and entinostat, (MS-275/SNDX-275; a Class I HDAC inhibitor) and checkpoint inhibitors improved treatment outcomes substantially, curing >80% of the tumour-bearing mice. The investigators revealed by functional studies that the main targets of the epigenetic modulators were myeloid-derived suppressor cells.
Conclusions and future directions
The treatment of MM with combination approaches has been described as entering a new era, where even target relapsed/refractory cases can be targeted.83,84 Increased understanding of genetic and epigenetic interactions in MM may ultimately facilitate the development of novel, individualised treatment strategies.63 Panobinostat and ricolinostat have shown efficacy in combination with bortezomib and dexamethasone in both newly diagnosed and heavily pre-treated patients with advanced MM. Panobinostat in combination with bortezomib and dexamethasone became the first HDACi approved for the treatment of MM, specifically for relapsed or RRMM in patients with ≥2 prior regimens including bortezomib and an immunomodulatory drug. HDAC inhibition in combination with lenalidomide and dexamethasone has proven effective even in patients who are refractory to lenalidomide, suggesting that panobinostat may be able to overcome drug resistance to IMIDS thus restoring lenalidomide sensitivity. The promising results in relapsed or RRMM from the PANORAMA 1 trial and other combination trials discussed here have paved the way to explore novel panobinostat combinations (panobinostat–carfilzomib or panobinostat–pomalidomide) as well as panobinostat combinations in other settings, such as front-line or maintenance therapy.
Preclinical evidence suggests that there is potential for HDACi–monoclonal antibody combinations in a clinical setting. There are also preclinical data supporting the basis for combining novel immunotherapeutics targeting programmed cell death protein 1 (PD-1/PD-ligand 1 pathway with HDACi.85 However, to progress this opportunity in a rational manner future preclinical work needs to focus on understanding the interplay between epigenetic regulation and the expression of antibody target antigens on MM cells and defining the immune modulatory capacity of HDACi in MM. This will inform the design of combinatorial therapeutic strategies to enhance monoclonal antibody-induced antibodydependent cell-mediated cytotoxicity in vitro and in vivo. There may also be an opportunity for a sequential therapeutic strategy to “prime” MM cells for monoclonal antibodies, chimeric antigen receptor-T cell therapy or other immune targeted therapies in development. While the prospects for enhancing MM outcome through the incorporation of HDACi into the therapeutic armamentarium are becoming evident it is clear that the optimal use of these agents requires further understanding of their pleiotropic anti-tumour capacity through future carefully designed preclinical and correlative studies.
Trial Names
PANORAMA 1 = Panobinostat or Placebo With Bortezomib and Dexamethasone in Patients With Relapsed Multiple Myeloma
PANORAMA 2 = Panobinostat in Combination with Bortezomib and Dexamethasone in Patients with Relapsed and Bortezomib-refractory Myeloma
VANTAGE = VorinostAt CliNical Trials in HemAtologic and solid maliGnanciEs