Myelodysplastic syndromes (MDS) are a group of clonal myeloid neoplasms characterised by blood cytopaenia, cellular dysplasia and predisposition to the development of acute myeloid leukaemia (AML).1,2 MDS are common in older individuals, with an incidence that varies from 2–12/100,000 inhabitants/year in the general population.3,4 The pathogenesis of MDS is due to acquired genetic changes that result in the development of an anomalous and genetically unstable stem cell clone.3 However, MDS may be classified as primary (de novo) or secondary, which can be associated with therapy.5 Primary MDS have advanced age as the main risk factor and represent the majority of cases, with chromosomal changes observed in 30–50% of cases.6 Secondary MDS develop after a known mutagenic event, such as exposure to radiotherapy or chemotherapy, and chromosomal alterations are present in up to 80% of cases.7
The diagnosis and prognosis of MDS can be can be determined by means of the Revised International Prognostic Scoring System (R-IPSS), which includes the evaluation of cytogenetic characteristics. Cytogenetic analysis also aids therapeutic decision making, facilitates the prediction of therapeutic response, and allows the identification of clonal evolution as a predictor of progression to AML in patients with MDS.8
The most frequent cytogenetic abnormalities in MDS involve cytogenetic losses resulting from monosomies or deletions, especially on chromosomes 5, 7, 20 or Y. Unbalanced translocations, as well as balanced structural abnormalities, rarely occur in MDS,9 and the isochromosome 7 [i(7q)] is a non-random cytogenetic abnormality, which is also rare in MDS.10 An isochromosome is an abnormal chromosome in which one of the arms is lost and the other is duplicated in the form of a mirror image. That is, an individual with i(7)(q10) has a 7p partial monosomy and 7q trisomy.11 Alteration i(7q) is closely associated with Shwachman–Diamond syndrome, Wilms’ tumour and hepatosplenic T-cell lymphoma.12 Cytogenetic abnormalities of chromosome 7
correspond to one of the most detected, and worst prognostic, changes in MDS. Specifically, the rare alteration i(7q), with undefined clinical and prognostic outcomes, especially in
young people. Therefore, we present the case of a young patient with MDS and clonal i(7)(q10) in bone marrow.
Case presentation
A 23-year-old male patient, was treated at the Hospital Universitário Walter Cantídio de Hematologia e Hemoterapia do Ceará hematology service, Fortaleza, Ceará, Brazil. At his first visit in 2012, he presented without symptoms with persistent thrombocytopaenia and leukopaenia. He was the only child of healthy and non-consanguineous parents. The following examinations were conducted: blood count, myelogram, bone marrow biopsy, immunohistochemical profile, immunophenotyping, karyotype analysis, and biochemical and serological studies for differential diagnosis. For the purpose of this case report, the results were collected retrospectively by analysis of medical records.
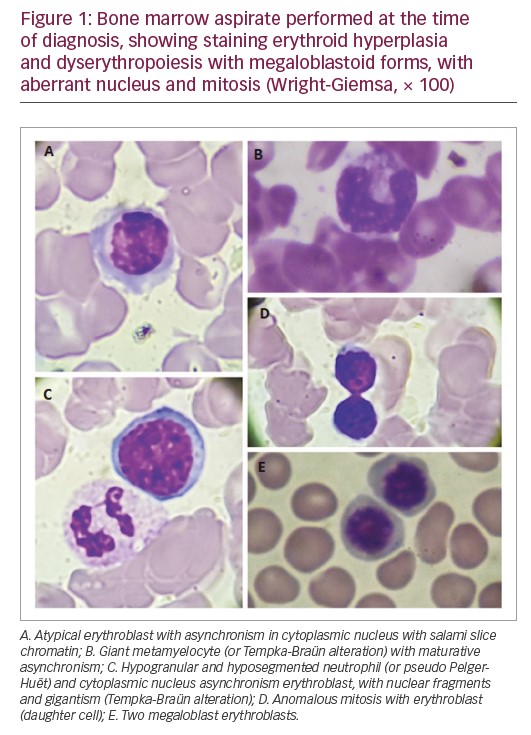
At diagnosis, the haemogram showed a red blood cell count of 3,950,000/mm³, haemoglobin of 13.3 g/dL, mean corpuscular volume of 103 f/L showing macrocytic and normochromic red blood cells, thrombocytopaenia (platelet count 68,000/mm³) and leukopaenia (white blood cell count 2,700/mm³). The myelogram revealed a relative erythroid hyperplasia and dyserythropoiesis with megaloblastoid forms with aberrant nucleus and mitosis figures (Figure 1). The granulocytic series revealed severe hypoplasia. Bone marrow histopathology revealed global hypocellularity with dysmegakarypoiesis, dyserythropoiesis, and absences of reticulinic and collagen fibrosis. Immunophenotyping showed a slight increase in vasculature, no increase in precursor cells, and negative reaction to P53. The immunohistochemical profile, associated with histopathological findings, was compatible with hypocellular bone marrow with mild lymphocytosis and megaloblastosis foci. There was positivity for CD34+, CD3+, CD20+, CD68+, LCA-CD25+, CD15+ myeloperoxidase, and negativity for CD30. JAK2 V617F and P53 mutations were negative.
To exclude other possibilities for cytopenic diagnosis, other laboratory results were analysed and were within the normal range: hepatic gamma-glutamyl transpeptidase 13 U/L, aspartate transaminase 31 U/L, alanine transaminase 33 U/L, total bilirubin 0.80 mg/dL, direct bilirubin 0.59 mg/dL, indirect bilirubin 0.61 mg/dL, increased alkaline phosphatase 527 U/L, lactate dehydrogenase 245 U/L. No renal dysfunction was reported and the following results were recorded: urea 26 mg/dL, creatinine 0.86 mg/dL, sodium 143 nmol/L, potassium 3.9 nmol/L. No solid neoplasms, thyroid dysfunction, autoimmune diseases or infections were reported (non-reagents for hepatitis B, hepatitis C and HIV); however, cytomegalovirus was detected (IgG 74 UA/mL). Differential diagnosis was negative for Shwachman–Diamond syndrome with no skeletal or pancreatic changes (fasting blood glucose <100 mg/dL).
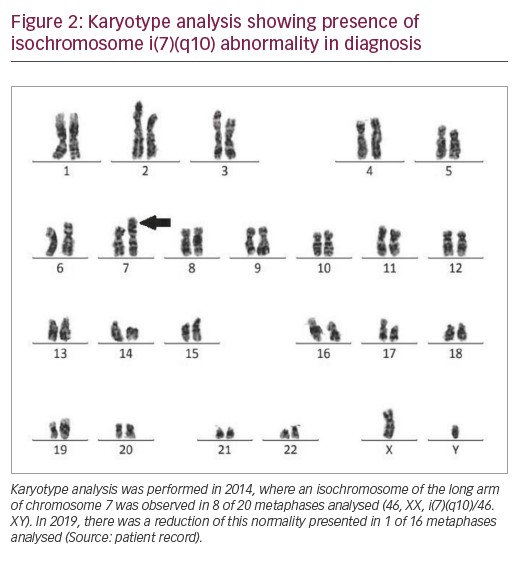
In 2014, the patient’s karyotype was analysed revealing isochromosome of the long arm of chromosome 7 in eight of 20 metaphases analysed (46, XX, i(7)(q10)/46.XY) (Figure 2). Thus, the patient was diagnosed with MDS with multilineage dysplasia with low cytogenetic risk by R-IPSS. The patient was monitored for 5 years, treated with folic acid, cyanocobalamin, pyridoxine and thiamine. He remained stable, i.e., without evolution to AML, and without the need for transfusion.
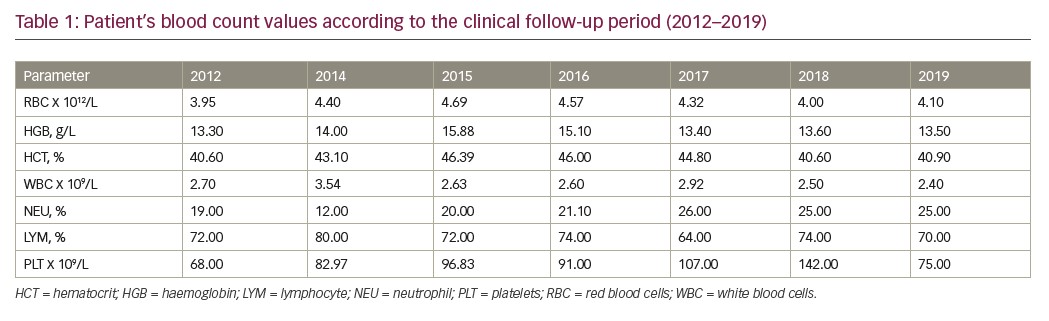
In 2019, a further karyotype analysis was performed, presenting isochromosome of the long arm 7 only in 1 of 16 metaphases analysed (46, XX, i(7)(q10)/46.XY). Blood count showed persistent thrombocytopaenia (platelet count 75,000/mm³), haemoglobin 13.6 g/dL, and leukocytes (white blood cell count 2,400/mm³). The clinical evolution of the patient from 2012–2019 is described in Table 1.
Discussion
Cytogenetic abnormalities are key in pathogenesis and diagnosis of MDS. Additionally, these abnormalities can be important prognostic indicators to assess survival, leukaemic transformation, and response to treatment.13 Cytogenetic abnormalities of chromosome 7 correspond to one of the most detected changes in MDS.14 Del(7q) and monosomy 7 are found in approximately 10% of MDS cases and are associated with lower survival.15 The isochromosome 7 [i(7q)] is a rare cytogenetic abnormality in MDS, especially in young people, and this is the first report of an individual with chromosomal abnormality i(7q) reported in north-eastern Brazil.
i(7)(q10) was first described to be associated with MDS/AML in 1996, due to pleiotropic effects of a mutant causal gene.16,17 Hypotheses regarding the mechanism of origin of i(7)(q10) include several factors, such as short repetitions in DNA near the centromere, instability of the spindle and defective telomeric function.13 It is believed that mutations in the SBDS gene, located in the 7q chromosomal arm, contribute to this cytogenetic alteration. The SBDS gene encodes a protein that promotes spindle stability, normal chromosomal segregation, and assembly of mature ribosomes.18 Therefore, mutations in this gene may either favour increased SBDS protein level, by providing an explanation for clonal expansion of the affected haematopoietic progenitor cell, or explain karyotypic instability, with acquired chromosomal changes.19
Generally, i(7)(q10) is observed in patients with Shwachman–Diamond syndrome, a hereditary syndrome which presents in childhood and includes cytopaenia, pancreatic failure, growth deficit, skeletal abnormalities, mental retardation and marrow failure.20,21 Interestingly, Shwachman–Diamond syndrome has a higher probability of progression to MDS/AML, due to karyotypic instability caused by mutations in the SBDS gene that are related to numerical and structural abnormalities of chromosome 7.22 According to the Severe Chronic Neutropenia International Registry, the incidence of MDS in individuals with Shwachman–Diamond syndrome is 8.1% over 10 years.23
The differential diagnosis of Shwachman–Diamond syndrome for MDS is based on the disease detection of pancreatic, hepatic, skeletal, and bone marrow changes.16 The presence of this abnormality in Shwachman–Diamond syndrome is associated with dysplasias, transfusion dependence and severe neutropaenia. In MDS, this change is associated with refractory cytopaenia with excess blasts or progression to AML.24 Based on this, the patient in this case report with cytogenetic alteration presents clinical manifestations related to MDS as persistent cytopenias without transfusion dependence. Studies suggest that in the absence of a diagnosis of Shwachman–Diamond syndrome, isochromosome 7q is considered as a primary alteration in the MDS/AML pathogenesis.22 However, the patient in the present case report presented with bone marrow alterations characteristic of MDS, such as megaloblastoid erythroblasts with asynchrony of nucleocytoplasmic maturation, erythroblasts with nucleus fragments, cellular gigantism (as the presence of giant metamielocytes, called Tempka-Braün alteration), hypogranular and hyposegmented neutrophils (Pelger–Huët) and other dysplasias, in which the diagnosis of Shwachman–Diamond syndrome was ruled out (Figure 1).
The presence of i(7)(q10) in patients with Shwachman–Diamond syndrome may persist for a long time or even regress, but may eventually progress to MDS/AML with increasing age. Babushok and Bessler believe that chromosomal changes, such as i(7)(q10) may remain stable in some cases and may or may not evolve from MDS to AML according to age.25 However, this relationship has been controversial in some studies, in which patients with clones i(7q) presented progressive bone marrow insufficiency with severe cytopaenia over time.13,20,26,27 Despite this, Leung et al. reported a case of transient MDS associated with i(7)(q10) in a child where, without intervention, bone marrow cytogenetics returned to normal and with complete hematologic recovery.24 In the present study, a new karyotype analysis was requested in 2019 to assess disease remission, showing a reduction in the percentage of abnormal metaphases.
The risk of MDS and the acquisition of bone marrow chromosomal changes are related to advancing age.13 Considering that MDS in young people is rare and assessment and follow-up are challenging, the need for clinical follow-up and cytogenetic analysis incorporated in the routine diagnosis of MDS to better determine the diagnosis, prognosis and treatment of patients with this disorder, is emphasised. Clinical investigation in patients with i(7)(q10) should consider the differential diagnosis of diseases such as Shwachman–Diamond, neoplasms, autoimmune diseases, immunodeficiency or infections, such as cytomegalovirus and the Epstein-Barr virus to define the best clinical management for the patient.
Although there is no consensus on the optimal treatment for patients with i(7)(q10), aggressive treatment with chemotherapy and haematopoietic stem cell transplantation is not indicated in patients with this abnormality.24 In the present case, the use of vitamins was an initial treatment attempt, but there was no response and was therefore discontinued. The patient maintained a stable clinical picture with persistent thrombocytopaenia requiring no transfusions or treatment with hypomethylating agents. Therefore, studies evaluating the pathogenesis of i(7) in MDS, and its association with prognosis, are relevant for better clarification of the disease.
Conclusion
Chromosomal abnormality, i(7)(q10) suggests a more favourable prognosis, since the number of metaphases with chromosomal abnormality decreased over time, remaining stable only with supportive treatment. However, the need for cytogenetic analysis in the routine diagnosis of MDS is highlighted, and further studies are required to better understand the clinical evolution of patients with this alteration.