Myelodysplastic Syndromes and 5q Deletion
Myelodysplastic syndromes (MDS) are a group of rare blood disorders that occur as a result of abnormal development of blood cells within the bone marrow. MDS may progress to life-threatening failure of the bone marrow or develop into an acute leukaemia. It affects, most commonly, the elderly population, with median age at diagnosis of about 65 years. MDS are a heterogeneous group of malignant haematopoietic stem cell disorders, which are characterised by dysplasia and ineffective production of blood cells, and increased risk of transformation to acute myeloid leukaemia (AML). Patients with MDS have variable cytopenias, i.e. reduction in the production of normal red blood cells (RBCs), platelets and white blood cells (mainly neutrophils). This may present as anaemia, bleeding and an increased risk of infection.1,2
The incidence of MDS in the US is estimated to be about 4.1/100,000 inhabitants/year. The incidence rises with increasing age: 4.9 for people aged 50 to 70 years; 22.8 for people older than 70 years.3 The incidence of MDS in Europe is approximately 4/100,000 inhabitants/year (increasing to 40–50/100,000 in patients aged ≥70 years). While there are no ethnic differences in the incidence of MDS, Asians tend to have MDS at an earlier age, more often show a hypo cellular marrow and less commonly present with isolated 5q deletion (5q- syndrome), while more commonly present with trisomy 8 compared with Western populations.4
The International Prognostic Scoring System (IPSS), which is the most commonly used tool for MDS, defines four prognostic risk categories: good, intermediate-1, intermediate-2 and poor, based on the number of cytopenias, proportion of marrow blasts and cytogenetic abnormalities.5 The revised version of IPSS defines five prognostic risk categories: very low, low, intermediate, high and very high, by further refining the same prognostic variables.6
Cytogenetic abnormalities can be detected in bone marrow cells in approximately 30–50 % of MDS patients. The most common chromosomal abnormalities seen in MDS are deletion of the long arm of chromosome 5 (del[5q]), -7 or del(7q), trisomy 8, del(20q) and loss of the Y chromosome.7 del(5q) has been reported to be present in approximately 10–15 % of all MDS cases either as the sole karyotype abnormality or as part of a more complex karyotype and it has distinct clinical implications for MDS.8
‘MDS associated with isolated del(5q)’ is recognised by The World Health Organisation (WHO) as a distinct entity,9 and it is characterised by an isolated del(5q), female predominance, macrocytic anaemia, <5 % bone marrow blasts, <1 % of peripheral blood blasts, a normal or increased platelet count and hypolobated megakaryocytes and no Auer rods. Patients with isolated del(5q) MDS have a stable clinical course and rare progression to AML. It has a relatively good prognosis and good chance of responding to treatment with lenalidomide.10 In comparison, del(5q) or structural rearrangements of 5q with other chromosomal changes lead to higher-risk MDS and worse outcomes.11
Profile of Lenalidomide
Lenalidomide, a thalidomide analogue, is an orally administered immunomodulator with anti-angiogenic, antineoplastic, anti-inflammatory and pro-erythropoietic properties.12 Lenalidomide was developed in effort to avoid thalidomide side effects, and to increase efficacy. Lenalidomide shares a number of structural and biological properties with thalidomide but it is safer and more potent than thalidomide.13
The key therapeutic mechanisms of action of lenalidomide include stimulation of erythropoiesis, immunomodulatory effects and antineoplastic effects, such as anti-angiogenic and antiproliferative activity. Immunomodulatory effects of lenalidomide mainly include: increasing the number and activity of T-cells, natural killer (NK) and NK T-cells; inhibition of the pro-inflammatory cytokines, such as tumour necrosis factor (TNF)-α; and, increasing the production of the anti-inflammatory cytokine interleukin (IL)-10 in peripheral blood mononuclear cells.14,15
In del(5q) MDS, lenalidomide causes a cytotoxic effect by stabilisation of the mouse double minute 2 homolog (MDM2) gene, and thereby leading to accelerated p53 degradation.16 In MDS with non-del(5q), lenalidomide acts by modifying bone marrow microenvironment, which abolishes the function of pro-apoptotic and pro-inflammatory cytokines an increased expression of adhesion and restoring erythropoiesis via erythropoietin (EPO)-induced activation of signal transducer and activator of transcription (STAT5) gene.17,18 Lenalidomide inhibits angiogenesis by inhibiting the migration and adhesion of endothelial cells induced by cytokines, such as vascular endothelial growth factor and basic fibroblast growth factor. In vitro, lenalidomide inhibits the expression of cyclooxygenase (COX)-2, but not COX-1, leading to reduction in prostaglandin E2 levels.14,15,19
Therapeutic Efficacy of Lenalidomide in Myelodysplastic Syndromes
Lenalidomide was first studied in a single-centre 16-week, phase I/II trial.20 This study included 43 patients with MDS and who had transfusion-dependence or symptomatic anaemia. All patients were refractory to EPO or had high endogenous EPO levels. Lenalidomide was administered in three different dosing schedules: 25 mg daily, 10 mg daily and 10 mg/day for 21 days of a 28-day cycle. Results of this study showed the potential of lenalidomide in EPO-resistant del(5q) MDS as well as the role of lenalidomide in EPO-refractory non-del(5q) MDS patients. Eighty-three per cent of patients with del(5q) MDS showed major erythroid responses, defined as sustained transfusion independence, compared with 53 % of patients with normal karyotype and 12 % of patients with other karyotype abnormalities. Dosedependent myelosuppression (neutropenia, thrombocytopenia) was the most common adverse event (AE); well-tolerated dose was 10 mg daily for 21 days of each 28-day cycle.
Lenalidomide in Myelodysplastic Syndromes Associated with del(5q) – Landmark Trials
Based on results of study mentioned above, the clinical efficacy of lenalidomide in patients with MDS associated with del(5q) was evaluated in two registration trials: the non-comparative, phase II, MDS-003 trial (n=148; US registration trial)21 and the phase III, double-blind, placebo-controlled, multicentre, MDS-004 trial (n=205) conducted in Europe and Israel.22
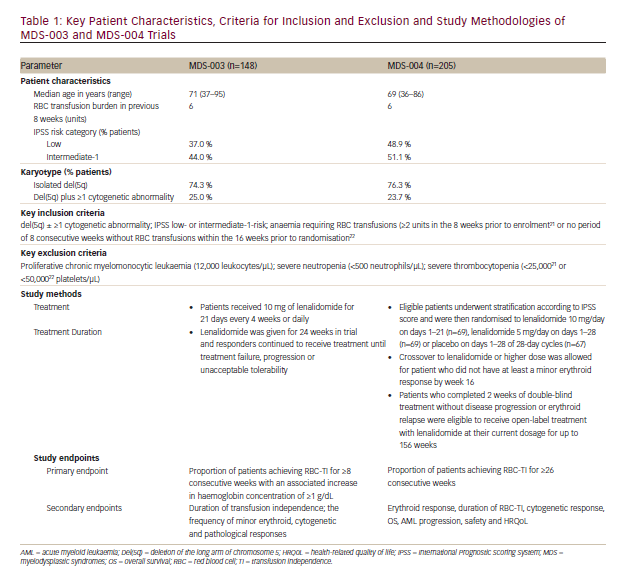
These landmark trials21,22 included adult patients with IPSS low- or intermediate-1-risk (lower risk) MDS associated with del(5q), with or without additional cytogenetic abnormalities. Included patients had RBC transfusion-dependent (TI) anaemia. Key patient characteristics, criteria for inclusion and exclusion and study methodologies are given in Table 1.
Results
Erythroid Response
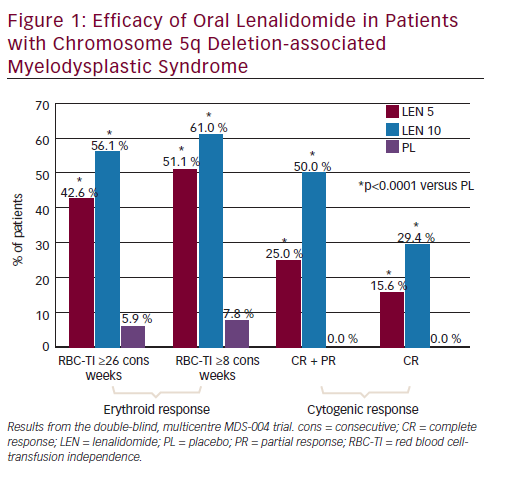
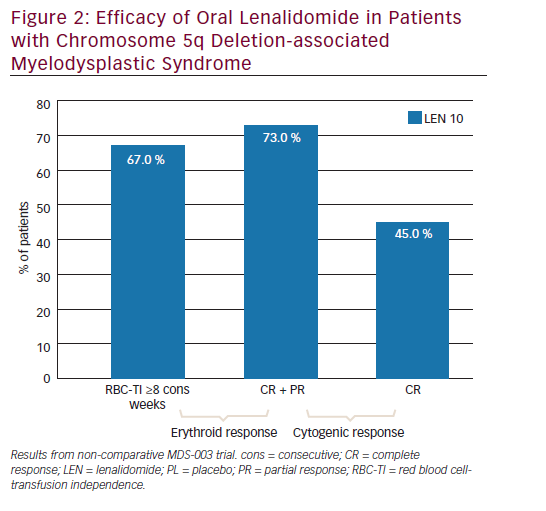
In the MDS-004 trial,22 a significantly higher proportion of patients in lenalidomide 10 and 5 mg groups experienced RBC-TI for >26 weeks versus placebo (56.1 % and 42.6 %, respectively, versus 5.9 %; both p<0.001) (see Figure 1). While in the MDS-003 trial, 67 % of patients receiving lenalidomide 10 mg/day achieved RBC-TI for ≥8 consecutive weeks and an associated ≥1 g/dL increase in haemoglobin level (see Figure 2).21
Subgroup Analysis of Erythroid Response
In MDS-004,22 subgroup analysis suggested that erythroid response (RBC-TI for ≥26 week) rates favoured lenalidomide 10 mg over 5 mg for most subgroups. Particularly, in patients with baseline EPO levels >500 mIU/mL, the RBC-TI rate was significantly higher with lenalidomide 10 mg versus 5 mg (76.2 % versus 33.3 %; p=0.004).
Time to Achieve Erythroid Response
Erythroid responses to lenalidomide treatment were achieved rapidly. In the MDS-004 trial, in patients who achieved RBC-TI for ≥26 consecutive weeks with lenalidomide treatment, the onset of response occurred in 86 % of responders during cycle one (48.8 %) or cycle two (37.2 %).22 This finding was consistent with that observed in the MDS-003 trial, in which the median time to an erythroid response was 4.6 weeks.21
Increase in Haemoglobin
In MDS-00321, with lenalidomide 10 mg/day treatment, haemoglobin concentration increased from a median minimum of 7.8 g/dL during the baseline period to a median maximum of 13.4 g/dL during RBC-TI response period, with a corresponding median rise from baseline of 5.4 g/dL. In MDS-004,22 the median increase from baseline (value not reported) in maximum haemoglobin concentrations were 5.2 and 6.3 g/ dL with lenalidomide 5 and 10 mg regimens, respectively.
Duration of Red Blood Cell Transfusion Independence
The median duration of RBC-TI for ≥8 consecutive weeks was not reached in either of these trials, and in MDS-004, the median duration of RBC-TI for ≥26 consecutive weeks was also not reached.22
Also, 60–67 % responders remained transfusion free at median followup of 1.55 years.
Cytogenetic Response
In MDS-004,22 treatment with lenalidomide 5 or 10 mg regimens resulted in significantly higher cytogenetic response rates than placebo (see Figure 1). With lenalidomide 10 mg regimens, complete plus partial cytogenetic responses were achieved by 50 % and 73 % of patients and a complete cytogenetic remission was seen in 29 % and 45 % of patients in the MDS-00422 and MDS-00321 trials, respectively (see Figures 1 and 2).
Disease Progression and Overall Survival
In the MDS-004 trial,22 after median follow-up of 30.9–36.1 months (double-blind and open-label treatment periods), the proportion of patients progressed to AML during the were 23.2 % and 21.7 % of patients receiving lenalidomide 5 or 10 mg regimens, 30.4 % of those randomised to placebo who crossed over to lenalidomide 5 mg/day
and 36.4 % of those receiving placebo throughout the study. Overall risk of AML progression for the lenalidomide dose groups combined was 16.8 % (95 % confidence interval [CI], 9.8-23.7) at 2 years and 25.1 % (95 % CI 17.1-33.1) at 3 years.
At a median follow-up of 35.5–36.9 months, median overall survival (OS) duration was ≥35.5 (95 % CI 24.6 – not reached), 44.5 (95 % CI 35.5 – not reached) and 42.4 (95 % CI 31.9 – not reached) months with lenalidomide 5 and 10 mg regimens and placebo, respectively.22
A retrospective analysis by Kuendgen et al.,23 compared long-term outcomes in lenalidomide-treated and untreated patients with MDS with del(5q). This analysis used data of 295 lenalidomide-treated patients from two clinical trials (MDS-003 and MDS-004) and 125 untreated and RBC-transfusion-dependent del(5q) MDS patients (IPSS low- or Intermediate-1-risk category) from a large multicentre registry. The 2-year cumulative AML progression rates (95 % CI) in the lenalidomide-treated versus untreated cohorts were 6.9 % (93.3–13.9) and 12.1 % (7.0–20.3). Two-year OS probabilities (95 % CI) were 89.9 % (84.1–96.0) and 74.4 % (66.1–83.7), respectively. AML progression risk did not significantly differ in both groups (hazard ratio [HR] 0.969, p=0.930); however, treatment with lenalidomide led to significant improvement in survival (HR 0.597; p=0.012), after adjusting for all other covariates. Thus, lenalidomide treatment did not increase the risk of AML progression and conferred a possible survival benefit in RBC transfusion-dependent patients with lower risk del(5q) MDS.
Health-related Quality of Life
Health-related quality of life (HRQoL) was assessed in the MDS-004 trial, using the patient-reported Functional Assessment of Cancer Therapy- Anemia (FACT-An) questionnaire. After 12 weeks of treatment (before non-responders were crossed over to lenalidomide) mean change of FACT-An total scores from baseline was significantly improved following treatment with lenalidomide 5 and 10 mg (+5.7 and +5.7, respectively) versus placebo (-2.8) (both p<0.05). Clinically important changes in HRQoL from baseline were observed at weeks 12, 24, 36 and 48 among RBC-TI ≥26 week responders in both lenalidomide groups.24,25
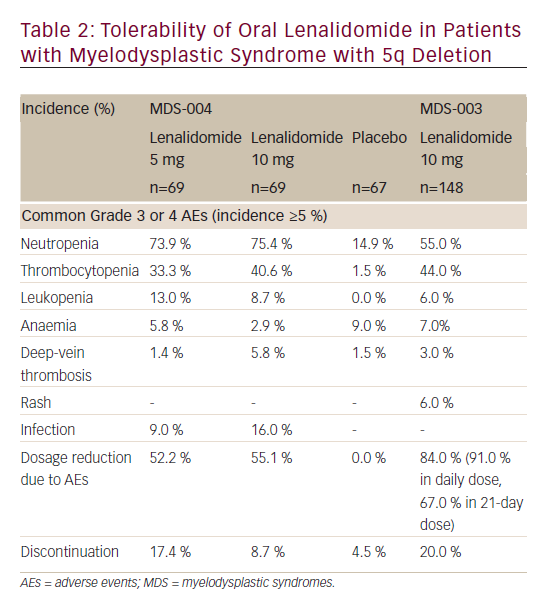
Tolerability
Oral lenalidomide (up to a dose of 10 mg/day) has a manageable safety profile in patients with del(5q)- MDS (see Table 2).21,22 Although many patients on lenalidomide experienced at least one grade 3 or 4 AEs in clinical trials, most were manageable with a dosage reduction and only ≤20 % of patients discontinued treatment because of AEs.
In the MDS-004 or MDS-003 trial, the most commonly reported (incidence ≥10 %) grade 3 or 4 AEs with lenalidomide treatment were neutropenia, thrombocytopenia, leukopenia and infection. Most common adverse reactions (>15 %) include thrombocytopenia, neutropenia, diarrhoea, pruritus, rash, fatigue, constipation, nausea, nasopharyngitis, arthralgia, pyrexia, back pain, peripheral oedema, cough, dizziness, headache, muscle cramp, dyspnoea, pharyngitis and epistaxis. Grade 3 or 4 neutropenia and thrombocytopenia occurred more frequently during the initial two cycles of lenalidomide treatment and the incidence decreased with further cycles.
Lenalidomide in Non-del-5q Myelodysplastic Syndromes
MDS-002 was a multicentre, phase II trial evaluating lenalidomide therapy for transfusion-dependent patients with low- or int-1–risk (lower risk) MDS without any or with non-del(5q) chromosomal abnormalities (n=214).26 Patients received oral lenalidomide 10 mg daily or 10 mg/ day for 21 of a 28-day cycle. The most common grade 3/4 AEs were neutropenia (30 %) and thrombocytopenia (25 %).
According to an intention-to-treat analysis, 26 % patients achieved TI after a median of 4.8 weeks of treatment. Median duration of TI was 41.0 weeks. In patients who achieved TI, the median rise in haemoglobin was 3.2 g/dL (range, 1.0–9.8 g/dL) from baseline. Overall rate of haematological improvement (HI) (TI response + ≥50 % reduction in transfusion requirement) was 43 %. Lenalidomide was found to have clinically meaningful activity in transfusion-dependent patients with lower-risk non-del-5q MDS.
MDS-005 was a multicentre, double-blind, parallel-group international phase III study, which compared lenalidomide with placebo in RBCtransfusion dependent lower-risk MDS without del(5q) patients, who were unresponsive or refractory to EPO-stimulating agents (ESA).27 Patients were randomised in 2:1 ratio; to oral lenalidomide, 10 mg once daily (n=160) versus placebo (n=79). The primary endpoint was RBC-TI of ≥56 days, which was defined as absence of any RBC transfusions during any 56 consecutive days. Secondary endpoints were time to TI, duration of TI, TI of ≥168 days, progression to AML, OS and safety. Significantly more patients on lenalidomide achieved RBC-TI ≥56 days versus placebo (26.9 % versus 2.5 %; p<0.001). Ninety per cent of patients with TI responded within 16 weeks of treatment. For patients who became transfusion independent, the median duration of TI was 8.2 months (range 5.2–17.8). Additionally, RBC-TI ≥168 days was achieved in 17.5 % in the lenalidomide group and 0 % of patients in placebo group. The incidence of AML progression (per 100 person–years) was 1.91 (95 % CI 0.80–4.59) and 2.46 (95 % CI 0.79–7.64) for lenalidomide and placebo patients, respectively. The follow-up period was not long enough to analyse OS comparison. In the study, the most common AEs associated with lenalidomide were related to myelosuppression. Grade 3/4 neutropenia occurred in 61.9 % and 12.7 % in the lenalidomide and placebo groups, respectively, and grade 3/4 thrombocytopenia occurred in 35.6 % versus 3.8 %. This study provided new insights into the clinical results of lenalidomide in non-del-5q patients. Lenalidomide may offer this refractory patient population an additional option beyond their current limited choices.
Combination Therapy in Higher Risk Myelodysplastic Syndromes
With lenalidomide’s success in lower-risk del(5q) MDS, it was further investigated for the treatment of higher-risk MDS. However, due to modest activity of lenalidomide monotherapy in higher-risk MDS, studies on lenalidomide in combination with other agents were required.
Thus, research was initiated with lenalidomide and azacitidine combination in MDS, since they work on different targets involved in the disease pathogenesis. In view of the single-agent activity of azacitidine and lenalidomide in MDS and AML, it was hypothesised that the combination of these two agents could potentially improve outcomes over monotherapy with either agent alone. Lenalidomide as immunomodulatory agent targets the microenvironment, while azacitidine works on DNA and RNA methylation. A multicentre phase II trial evaluated the use of combination azacitidine at 75 mg/m2 for 5 days with lenalidomide 10 mg/day for 21 days of 28-day cycle.28 This trial specifically targeted higher-risk MDS patients; total 36 patients: five patients IPSS intermediate-1, 20 patients IPSS intermediate-2 and 11 patients IPSS high. Overall response rate was 72 %, out of which 44 % complete response (CR) and 28 % with HI. Median OS was over 37 months for CR and 13.6 months for the entire study population. Commonly reported AEs were febrile neutropenia (22 %), infection (11 %), pulmonary AE (11 %), cardiac AE (11 %), constitutional (11 %) and dermatological AE (11 %). Thus, lenalidomide/azacitidine combination was found to be welltolerated and highly active in treatment of higher-risk MDS.28
A phase II trial by Sekeres et al. compared the addition of vorinostat or lenalidomide to azacitidine, with azacitidine alone. This study included 276 evaluable patients with higher-risk MDS, defined as intermediate-2 or high-risk IPSS score and/or bone marrow blasts ≥5 % (n=226), as well as patients with chronic myelomonocytic leukaemia (CMML) and <20 % blasts (n=50).29 Patients were randomised to one of three treatment arms: azacitidine 75 mg/m2 on days 1–7 of each 28-day cycle (n=92); azacitidine plus lenalidomide 10 mg/day on days 1–21 (n=93); or azacitidine plus vorinostat 300 mg twice daily (BID) on days 3–9 (n=91). The overall response rates were similar for all patients regardless of treatment arm; single-agent azacitidine (36 %), azacitidine plus lenalidomide (37 %; p=1.0 versus azacitidine monotherapy), or azacitidine plus vorinostat (22 %; p=0.07 versus azacitidine monotherapy). Also, the proportion of patients achieving CR, partial response (PR) and HI were similar across all treatment groups. Thus, compared with azacitidine alone, the combination of vorinostat or lenalidomide with azacitidine failed to improve response in patients with higher-risk MDS and CMML.
The dose and schedule for azacitidine-lenalidomide combination used by Sekeres et al. might not be optimum.29 As lenalidomide inhibits cell cycle progression and effectiveness of azacitidine is dependent on cycling cells, sequential administration of azacitidine followed by lenalidomide might increase efficacy. To study such a schedule, DiNardo et al.30 conducted a phase I/II trial of the sequential combination of azacitidine followed by lenalidomide in high-risk patients with MDS and AML. During phase I and IIa, patients with relapsed or refractory AML or MDS with bone marrow blasts more than 10 % were included. Phase IIb included previously untreated patients with higher-risk MDS with up to 30 % blasts. All participants received 75 mg/m2 azacitidine once a day for days 1–5 for each 28-day cycle. Oral lenalidomide was given for 5 or 10 days starting on day 6 in seven different doses in phase I design (n=28). In total 88 patients were enrolled, 28 in phase I and 60 in phase II. A maximum tolerated dose of lenalidomide in combination with azacitidine was initially considered as 50 mg per day for 10 days. However, due to the high rate of myelosuppression and myelosuppression-related toxic effects in the first 20 patients in phase II, the lenalidomide dose was revised to 25 mg per day for 5 days. Also in phase II patients with less than 30 % blasts were included to focus mainly on MDS. Median OS was 75 weeks for the 40 patients in phase IIb. The most common grade 3–4 AEs overall were neutropenic fever and pneumonia. Thus, this study found a safe sequential combination strategy for azacitidine and lenalidomide for patients with higher-risk MDS, 75 mg/m2 intravenous azacitidine once a day for 5 days followed by 25 mg oral lenalidomide once a day for 5 days. Also, this study provided preliminary evidence of activity of this schedule in patients with MDS.
Lenalidomide – Summary and Current Status for its Use in Myelodysplastic Syndromes
Lenalidomide is a novel thalidomide analogue with enhanced immunomodulatory and anti-angiogenic action lacking most of the typical thalidomide-associated AEs. Oral lenalidomide is approved in the US for the treatment of patients with transfusion-dependent anaemia due to lower risk (i.e. IPSS low- or intermediate-1-risk) del(5q) MDS, with or without additional cytogenetic abnormalities.31 While in Europe, it is indicated for the treatment of patients with transfusiondependent anaemia due to lower-risk MDS associated with an isolated del(5q) cytogenetic abnormality when other therapeutic options are insufficient or inadequate.32
According to the US National Comprehensive Cancer Network (NCCN) guidelines, patients with lower-risk del(5q) MDS who have clinically significant cytopenias and symptomatic anaemia should be treated with lenalidomide, along with supportive care as an adjunct to the treatment (category 2A recommendation).33 Lenalidomide is also an option for patients with lower-risk non-del(5q) MDS, who have clinically significant cytopenias and symptomatic anaemia, with either a serum EPO level of >500 mU/mL and a poor probability of responding to immunosuppressive therapy, or a serum EPO level of ≤500 mU/mL and who have not responded to ESA (category 2A recommendation).33 Guidelines from the European Society for Medical Oncology (ESMO) indicate that patients with 5q-syndrome may be treated with lenalidomide.4
The clinical efficacy of lenalidomide in patients with lower-risk del(5q) MDS observed in the phase II MDS-003 trial was further confirmed in the phase III MDS-004 trial. Treatment with lenalidomide in this population led to RBC-TI for ≥26 consecutive weeks and cytogenetic responses in a significantly higher proportion of patients than placebo. Erythroid responses are achieved rapidly and about 60–67 % of responders remained transfusion free at a median follow-up of 1.55 years. Lenalidomide generally has a manageable tolerability profile. The most common AEs occurring in lenalidomide recipients are thrombocytopenia and neutropenia, most cases can be managed by dosage reductions. Thus, lenalidomide is a useful option for the treatment of patients with lower-risk del(5q) MDS, with or without additional cytogenetic abnormalities. Recently published results of MDS-005 also suggested its role also in lower-risk MDS without del(5q).27 Recent trials have focused on combining lenalidomide with other agents active for treatment of higher-risk MDS.29,30
There are currently no sufficient data elucidating long-term benefits of lenalidomide therapy. Moreover, it remains unclear to decide when it can be discontinued. Further studies investigating the role of lenalidomide in lower-risk MDS without del(5q) and its role in higher-risk MDS in combination with other agents are warranted, which can give more clarity on the dosing schedule and efficacy of such combinations. Studies assessing the impact of long-term immunomodulation with lenalidomide and optimum duration of therapy in MDS patients would also be of interest.