Multiple myeloma (MM) is the second most common hematologic malignancy representing 1.6 % of all new cancer cases in the US. The rates for new MM cases have been rising on an average of 0.8 % each year over the last 10 years. While the survival of patients with MM has steadily improved over the last decade, nearly all patients with the disease relapse and eventually succumb to refractory disease. The 5-year survival is 46.6 % as per Surveillance, Epidemiology, and End Results (SEER) analysis.1
The incorporation of novel drugs, particularly thalidomide (Thal), lenalidomide (Len), and bortezomib (Btz) has resulted in significant prolongation of overall survival (OS) in newly diagnosed as well as relapsed patients.2 The results with these novel agents during induction and in consolidation/maintenance post autologous stem cell transplant (ASCT) have also been encouraging.2,3
Although MM has become a more treatable disease, the development of resistant/refractory disease is inevitable, leading to fatal outcomes in the vast majority of patients. Therefore, there is an urgent need for therapies that can further extend disease control of these patients, without addition of significant treatment-related toxicities. This led to the exploration of monoclonal antibodies (MoAbs) in MM, which offer a completely different mechanism of action and based on the results seen in other cancers, may have significant efficacy in MM. However, despite the impressive efficacy of the anti-cluster of differentiation 20 (CD20) MoAb, rituximab, in the treatment of B-cell non-Hodgkin’s lymphoma, early studies of anti-CD20 rituximab and anti-interleukin-6 (IL-6) antibodies in MM have shown only minimal activity.4–6 Since then a large variety of MoAbs directed against numerous antigen targets on myeloma cells or on components of the bone marrow (BM) microenvironment (BM stromal cells [BMSCs] or signaling molecules) have been evaluated.
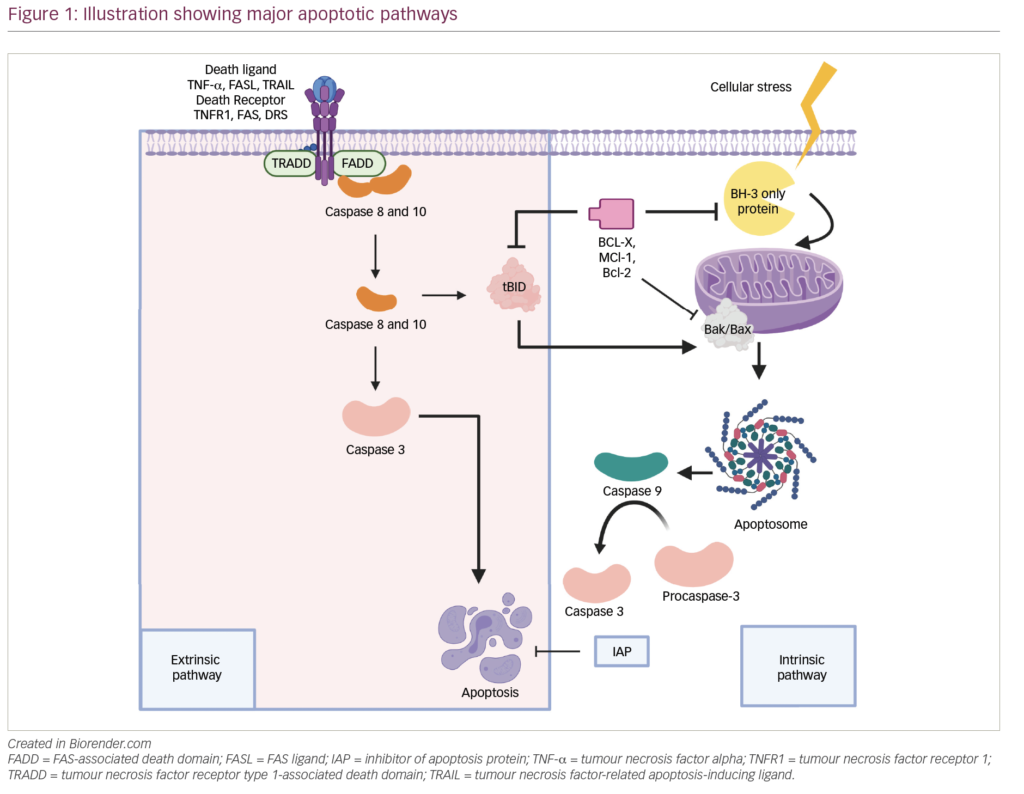
Most of the approved therapeutic MoAbs belong to the immunoglobulin-G1 (IgG1) subclass, which has a long half-life and activates immune effector functions. After the binding of MoAbs to a specific target on tumor cells antibody-dependent cellular cytotoxicity (ADCC) or complement dependent cytotoxicity (CDC) is triggered, thereby killing the target cell. Also, MoAbs could directly induce apoptosis or growth arrest upon binding to a cell surface antigen on the tumor cells. Other possible mechanisms include antibody dependent phagocytosis, interference with ligand binding, and the use of MoAbs as carriers of cytotoxic agents..7 .The potential targets include signaling molecules, cell surface receptors,plasma cell growth factors, and mediators for adhesion of the myeloma cells to the cells in the BM microenvironment..8.
This review focuses on the use of MoAbs alone or in combination with other agents in the therapy of MM, focusing on agents demonstrating high level of efficacy in preclinical and clinical studies. The most promising MM-related antigen targets and their corresponding MoAbs in various phases of preclinical and clinical development are listed in Tables 1–3.
Antibodies Targeting Cell Surface Molecules
Signaling Lymphocyte Activating Molecule-F7
Signaling lymphocyte activating molecule-F7 (SLAMF7) (CS1, CD2 subset 1, CRACC, CD319, or 19A24) is a cell surface glycoprotein of the SLAMrelated receptor family, with high expression on the surface of myeloma cell lines and CD138-purified primary tumor cells from MM patients. It is also expressed on natural killer (NK) cells, a subset of T cells, activated monocytes, and mature dendritic cells, but not on normal tissues and CD34 positive stem cells..9. The role of SLAMF7 is not well understood; however, it may contribute to MM pathogenesis by increasing myeloma cell adhesion and clonogenic growth via c-maf-mediated interactions with BMSCs9 and preserving myeloma cells from apoptosis by lowering phosphorylation of extracellular signal-regulated kinase (ERK) 1/2, Akt, and signal transducer and activator of transcription (STAT).10.
Elotuzumab (Elo) is a humanized MoAb against SLAMF7 that has significant antimyeloma activity both in vitro and in vivo.9,11 The preclinical studies demonstrated that the anti-myeloma activity of Elo is induced by ADCC mediated by NK cells. Moreover, it triggered autologous ADCC against primary myeloma cells resistant to conventional or novel therapies including Btz and HSP90 inhibitors, and considerably enhanced HuLuc63- induced myeloma cell lysis when pretreated with conventional or novel anti-MM drugs. Specifically, the combination of Elo with Btz significantly increases the in vivo therapeutic efficacy to eradicate patient derived myeloma cells in a severe combined immunodeficiency mouse-human (SCID-hu) mouse model.12
In a phase I, multicentric, dose-escalation study of Elo in patients with relapsed/refractory MM (RRMM), the adverse events were generally mild to moderate in severity and included cough, headache, back pain, fever, and chills. However, its antitumor activity was only modest with no responses and only 26.5 % of patients achieving stable disease (SD).13
Despite the limited activity of Elo monotherapy, subsequent clinical studies evaluated its combination with cytotoxic chemotherapy and immunomodulatory drugs. In a phase I study of Elo plus Btz in RRMM, an objective response rate (ORR) of 48 % was observed in 27 evaluable patients and in two out of three patients refractory to Btz. The median time to progression in these patients was 9.5 months.14 Jakubowiak et al. randomized patients with RRMM to Btz/dexamethasone (Dex) with or without Elo. He reported that the addition of Elo to Btz/Dex leads to an improvement in median progression-free survival (PFS) from 6.9 months to 9.7 months (p=0.08).15
In another phase II study evaluating Elo in combination with Len and lowdose Dex in RRMM, the ORR was 84 %. It was also observed that Elo at 10 mg/kg resulted in a high ORR and median PFS compared with the 20 mg/kg dose.16 A phase III study (ELOQUENT-2) randomized patients with RRMM to Len/Dex with or without Elo. Co-primary endpoints were ORR and PFS. Six hundred and forty-six patients were randomized (321 in the Elo with Len/Dex group; 325 in the control group). The patients had received a median of two prior lines of therapy (including Btz in 70 %, Thal in 48 %, and Len in 6 % of patients). Around 32 % of patients had deletion of 17p and 35 % were refractory to the last therapy. The ORR was 79 % in the Elo group versus 66 % in the control group (p<0.001). The median PFS increased from 14.9 months to 19.5 months (p<0.001) with the addition of Elo to Len/Dex.
Serious adverse events were reported in 65 % and 57 % of patients in the Elo and control group, respectively. Grade 3–4 adverse events were anemia (19 % versus 21 %), neutropenia (34 % versus 44 %), and lymphocytopenia (77 % versus 49 %) in the Elo versus control group. The exposure-adjusted infection rate was the same in both arms, but the rate of herpes zoster infection was greater in the Elo group. The authors concluded that the addition of Elo had a significant relative reduction of 30 % in the risk for disease progression or death.17 Hence, despite its lack of efficacy as a monotherapy, it appears to be promising in combination therapy.
ELOQUENT-1, a phase III trial, evaluating the role of addition of Elo to Len/Dex in patients with newly diagnosed MM is ongoing. The primary endpoint is PFS and the secondary endpoints are ORR and OS.18
Cluster of Differentiation 38
CD38 is a 46 kDa type II transmembrane glycoprotein that has a relatively high expression on all malignant myeloma cells and plays an important role in cell adhesion and signaling.19
There are three CD38 MoAbs in clinical development: a humanized MoAb (isatuximab) and two fully human MoAbs (daratumumab and MOR202). Daratumumab (HuMax®–CD38, Genmab, Copenhagen, Denmark) was synthesized after immunizing transgenic mice (HuMax®-Mouse) possessing human, but not mouse, Ig genes. Preclinical studies indicated that HuMax- CD38 was effective in killing primary CD38+ CD138+ patient myeloma cells and a range of myeloma/lymphoid cell lines by both ADCC and CDC.20 Daratumumab inhibits the CD38 adenosine diphosphate (ADP)-ribosyl cyclase activity in target cells, which may contribute to the effectiveness of daratumumab in killing both primary MM and plasma cell leukemia cells. Importantly, daratumumab induced ADCC and CDC were not affected by the presence of BMSC.21
In a phase I dose-escalation study of heavily pretreated RRMM, singleagent daratumumab showed a favorable safety profile. Overall, 42 % of this heavily pretreated population achieved a partial response (PR) or better at doses ≥4 mg/kg.22 Lokhorst et al. evaluated daratumumab in patients with RRMM in a phase I/II trial. Patients had received a median of four prior treatments. Seventy-nine percent of the patients were refractory to the last therapy received (64 % were refractory to proteasome inhibitors and immunomodulatory drugs; 64 % were refractory to Btz and Len), and 76 % had received ASCT. The most common grade 3 or 4 adverse events were pneumonia and thrombocytopenia. The ORR was 36 % and the median PFS was 5.6 months in the cohort that received 16 mg per kilogram.23 The results were remarkable considering the highly refractory nature of the patient population. It has since been found that Len potentiates daratumumab dependent ADCC in myeloma cell lines, following a study by Mutis et al. in which they added daratumumab to Len/Btz combination. They demonstrated that its addition significantly improved the effectiveness of Len and Btz treatment alone.24
Similarly, MOR202 (MorphoSysAG), a fully human anti-CD38 IgG1 MoAb produced by a human combinatorial antibody library (HuCAL) platform, also efficiently triggers ADCC against CD38+ myeloma cell lines and patient myeloma cells in vitro as well as in vivo in a xenograft mouse model.25 Hence, anti-CD38 MoAbs appear to be a promising option as monotherapy as well as the Len/daratumumab combination. However, one practical problem in applying anti-CD38 therapy is the wide expression on lymphoid, myeloid, and epithelial cells, especially following cell activation.
Syndecan-1 Syndecan-1
(CD138) is a heparan sulfate proteoglycan that serves as a receptor for epidermal growth factor (EGF) ligands. It is frequently present on normal and myeloma plasma cells and is involved in cell adhesion, maturation, and proliferation. When present at high levels in the serum, CD138 is an independent indicator of poor prognosis.26,27 Also, almost all myeloma cells, even after exposure to multiple therapies, express the antigen, making it a useful target at any stage of the disease.
Three new murine/human chimeric anti-CD138 antibody–maytansinoid conjugates; nBT062-SMCC-DM1, nBT062-SPDB-DM4, and nBT062-SPP-DM1 were found to have antitumor efficacy against myeloma cell lines and primary tumor cells from MM patients by inducing G2–M cell cycle arrest, followed by apoptosis associated with cleavage of caspase-3, caspase-8, caspase-9, and poly (ADP-ribose) polymerase. They vary in their linkage between the maytansinoid moiety and MoAb. Nonconjugated nBT062 completely block cytotoxicity induced by nBT062–maytansinoid conjugate, confirming that specific binding is required for inducing cytotoxicity. Moreover, nBT062– maytansinoid conjugates block adhesion of myeloma cells to BMSC.28 Preliminary data from an ongoing phase I/II a study to evaluate the safety and efficacy of BT062 in combination with Len/Dex in patients with RRMM indicated that BT062 is well tolerated. It induced responses even in patients with prior exposure to both Len and Btz and patients refractory to prior Len.29
Also, Rousseau et al. reported the preliminary bio distribution and dosimetry results of iodine-131-labeled anti-CD138 MoAb (B-B4) in RRMM patients in a phase I/II radioimmunotherapy study. Four patients with progressive disease (PD) after three lines of therapy were enrolled. Grade 3 thrombocytopenia was documented in two cases, and no grade 4 hematologic toxicity was observed. One patient experienced PR, with 60 % reduction in monoclonal spike (M-spike), and total alleviation of pain, lasting for 1 year.30
Cluster of Differentiation 20
Based on the landmark success of rituximab in CD20-positive B-cell lymphomas, Zojer et al. studied rituximab in RRMM. However, despite significant reductions in circulating B cells and serum IgM levels, it did not achieve any beneficial clinical effect.31 Moreover, rituximab was also evaluated as maintenance therapy in MM following ASCT, but its use in this setting was associated with an unexpectedly high rate of early relapse. The authors hypothesized a possible role for rituximab in provoking an additional reduction in the normal, residual B-cell activity as the underlying mechanism.4
In most of these studies, patients were not selected according to the level of CD20 expression on plasma cells. The majority of these patients did not express CD20-positive phenotype at the time of therapy. So, Moreau et al. conducted a prospective phase II trial of four weekly infusions of 375 mg/m2 rituximab in patients with MM expressing CD20 on at least 33 % of tumor cells. A total of 14 patients were enrolled who had a median of 75 % tumor cells expressing CD20 at the onset of therapy. Rituximab was well tolerated in all the patients. One patient experienced a minor response and five patients experienced SD. The patient who achieved a minor response expressed CD20 in 100 % of its plasma cells at the time of rituximab therapy; however, the BM examination performed after 3 months of rituximab therapy showed only 0.6 % of plasma cells, none of them expressing CD20. Conversely, in three other cases, the percentage of CD20+ plasma cells did not decrease despite rituximab therapy.32
Taken together, the resistance of myeloma cells against rituximab could be multifactorial; including the level of CD20 expression, dissociated action of CDC and ADCC, polymorphism in Fc-γ receptor, and an inadequate dose schedule.33
By contrast, there have been other studies that demonstrated that the CD20+ phenotype is associated with t(11, 14) (q13; q32) cytogenetic abnormality and with shorter survival.34 Also, circulating clonotypic cells with properties similar to normal memory B cells have been found to have clonogenic potential in MM.35 Hence, it may be too soon to consider it ineffective and clinical trials using rituximab in MM deserve further investigation.
Cluster of Differentiation 40
CD40 is a transmembrane protein belonging to the tumor necrosis factor (TNF)-α superfamily, which is expressed in high levels on the surface of myeloma cell lines and primary myeloma cells.36 The binding of CD40 to its natural ligand leads to myeloma cell proliferation and migration via the phosphatidylinositol-3 kinase (PI3K)–Akt–nuclear factor kappa-lightchain- enhancer of activated B cells (NF-kB) signaling.37 CD40 is also expressed by BMSCs and upon activation it triggers the secretion of IL-6 and vascular endothelial growth factor (VEGF) pathway.38,39
Two MoAbs targeting CD40 activation in myeloma cells, lucatumumab (also known as HCD122) and dacetuzumab (also known as SGN-40), have been investigated. Lucatumumab is a fully human, recombinant IgG1 type anti- CD40. An open-label, multicenter, phase I study found that single-agent lucatumumab was well tolerated up to a dose of 4.5 mg/kg. It induced SD in 43 % and PR in 7 % of RRMM patients.40 Dacetuzumab is a humanized anti-CD40 MoAb. In a phase I study, it was found to be well tolerated, but did not produce any objective responses.41 However, preliminary in vitro data suggested that combining dacetuzumab with Len is synergistic.42 Hence, a phase I study using this combination in RRMM patients was conducted. The regimen was generally well tolerated with fatigue, neutropenia, and thrombocytopenia as the most common adverse events. It showed an ORR of 61 % in Len-naïve patients and 28 % in patients previously treated with Len.43
Cluster of Differentiation 56
CD56 is a membrane glycoprotein from the Ig superfamily, expressed on neurons, muscle cells, NK cells, and also expressed on 70–90 % of myeloma cells. It appears to mediate cell adhesion, migration, and invasion.44 Moreover, expression of CD56/neural cell adhesion molecule correlates with the presence of lytic bone lesions in MM and distinguishes myeloma from monoclonal gammopathy of unknown significance (MGUS) and lymphomas with plasmacytoid differentiation.45
Lorvotuzumab mertansine (also known as IMGN901) is an immunoconjugate composed of a humanized anti-CD56 MoAb (huN901) conjugated to maytansinoid N2‘-deacetyl-N2‘-(3-mercapto-1-oxopropyl)-maytansine (DM1), a potent antimicrotubular cytotoxic agent. The phase I clinical study of lorvotuzumab mertansine in 23 patients with MM determined the maximum tolerated dose (MTD) as 140 mg/m2/week dose and demonstrated an overall favorable safety profile. Three patients showed mixed response, eight patients showed SD, and two patients remained on treatment for 42 weeks.46 In a phase I study of lorvotuzumab mertansine in combination with Len/Dex in patients with CD56-positive RRMM, the responses were modest. The most common adverse event was peripheral neuropathy, which occurred more frequently at higher doses (55 % at 75 mg/m2 and 100 % at 90 and 112 mg/m2) and after three cycles.47 Owing to the synergistic activity with Len and Btz,48 phase I studies utilizing this combination are presently ongoing.
Cluster of Differentiation 74
CD74 expression has been demonstrated in more than 80 % of MM cases.49 Milatuzumab (also known as IMMU-110) is an immunoconjugate composed of doxorubicin conjugated to an anti-CD74 MoAb, LL1. A phase I multicentric study evaluated milatuzumab in patients with RRMM. Initially, infusion reactions were reported as the dose-limiting toxicity. After increasing prophylactic medications and slowing administration they were only of grade 1 or 2 intensity. However, there were severe adverse effects including bacterial meningitis, fever, unexplained drop in hemoglobin, cord compression, confusion, hypercalcemia, and thrombocytopenia. There were no objective responses, but 26 % of those who completed treatment in this heavily pretreated and generally refractory group had SD for ≥3 months.50
HM1.24
HM1.24 (also known as CD317) is an important activator of NF-kB pathway and plays a major role in trafficking and signaling between the intracellular and cell surface of myeloma cells.51 A phase I/II clinical study reported that a humanized anti-HM1.24 MoAb, AHM, did not cause any serious toxicity when administered to patients with RRMM.52 XmAb5592 is a novel Fc engineered and humanized anti-HM1.24 MoAb that has significantly enhanced binding to all human FcγR receptors and triggers 10–100-fold higher ADCC against these myeloma cell lines than a native/non-Fcengineered version.53
Others Potential Targets
Other potential targets are CD48, CD70, CD229, GM-2 ganglioside, programmed cell death protein-1 (PD-1), beta 2 microglobulin, insulin-like growth factor 1 receptor (IGF-1), human leukocyte antigen-DR (HLA-DR), and TRAIL signaling pathways, the antibodies against which are presently studied in various preclinical and clinical trials.54–63
Antibodies Targeting Cytokines in Tumor Microenvironment
The BM microenvironment encompasses a wide spectrum of cell types and extracellular matrix proteins, including fibronectin, collagen, laminin, and osteopontin. A cardinal clinical feature of MM is the presence of osteolytic bone lesions due to imbalance between bone formation and resorption. Various clinical studies have linked the level of bone disease with the myeloma disease burden.64–66 The inhibition of the wingless (Wnt) pathway suppresses osteoblasts, whereas the amplification of the receptor activator of nuclear factor-kappa B (RANK) pathway and the action of macrophage inflammatory protein-1-alpha (MIP1α) activates osteoclasts.64 The induction of proangiogenic molecules (e.g., VEGF) enhances the microvascular density of BM and accounts for the abnormal structure of myeloma tumor vessels.65 Hence, these cytokines and the downstream pathways appear to be an interesting target for immunotherapy.
Interleukin-6
IL-6 is a key cytokine predominantly produced by BMSCs. It mediates myeloma cell growth and prevents apoptotic cell death via at least three different signaling pathways: Ras/MEK/ERK cascade, janus kinase-2 (JAK2)/STAT-3 cascade, and PI3K/Akt cascade. Both IL-6 and its receptor, IL-6R, are potential targets for MoAb-based intervention.66
Siltuximab (also known as CNTO 328) is a chimeric anti-IL-6 MoAb. Preliminary results of siltuximab in combination with Btz showed an encouraging ORR of 57 %. But grade 3 or more hematologic toxicities were somewhat common.67 A phase II, randomized, double-blind study demonstrated that the combination of siltuximab and Btz lead to a nonsignificantly higher ORR (55 % versus 47 %) and median PFS (8.1 months versus 7.6 months) compared with placebo and Btz in RRMM. However there was no difference in OS.68 Also, Voorhees et al. demonstrated that siltuximab could enhance the apoptotic activity of Dex as evidenced by increased activation of caspase-8, caspase-9, and caspase-3, annexin V staining, and DNA fragmentation.69
Elsilimomab (also known as BE-8) is a murine anti-IL-6 MoAb. The addition of elsilimomab to Dex and high-dose melphalan (Mel) during conditioning in ASCT demonstrated a response in 81.3 % of patients (complete response [CR] in 37.5 % of patients).70 Hence, the evaluation of siltuximab in combination with Mel-based therapies appeared promising. A phase II randomized study of Btz/Mel/prednisone with or without siltuximab was conducted in transplant ineligible newly diagnosed MM. However, this study did not reveal any improvement in CR or long-term outcomes with addition of siltuximab.71
Tocilizumab is a humanized anti-IL-6 MoAb. It is currently approved for rheumatoid arthritis in several countries and for Castleman disease in Japan. Tocilizumab was reported to inhibit in vitro proliferation of cloned and freshly isolated myeloma cells from patients with advanced MM and is presently being evaluated in open-label phase I and II trials.72
Vascular Endothelial Growth Factor
In addition to its angiogenic effects, VEGF also functions as a growth factor for myeloma cells, stimulates IL-6 production in BMSCs, triggers myeloma cell migration, and inhibits maturation of dendritic cells. Elevated levels of VEGF have been associated with poor survival in MM patients.73
A phase II study of the bevacizumab with Len and pulse Dex in RRMM showed an encouraging ORR of 70 % in the 27 evaluable patients. However, these outcomes were was not statistically superior to the 60 % ORR achieved in previously treated MM patients who received Len and high-dose Dex.74 Also, the addition of bevacizumab to Btz was well tolerated in unselected patients with pretreated MM. But the combination did not result in significant improvements in efficacy outcomes.75
B-cell-activating Factor
B-cell-activating factor (BAFF) is a TNF superfamily member produced in the BM microenvironment and is recognized as new survival factor for MM.76 Binding of BAFF to the three TNF-R-related receptors transmembrane activator and calcium-modulator and cytophilin ligand interactor (TACI), B cell maturation antigen (BCMA), and BAFF-receptor (BAFF-R) triggers activation of NF-kB, PI3K, and mitogen-activated protein kinase (MAPK) pathways, resulting in survival and Dex resistance of myeloma cells.77,78
Atacicept acts as a decoy receptor by binding to and neutralizing soluble BAFF and a proliferation-inducing ligand (APRIL). It prevents these ligands from binding to their cognate receptors on B-cell tumors, thereby enhancing cytotoxicity. An open-label, dose-escalation, phase I/II study of atacicept in RRMM or active, progressive Waldenstrom macroglobulinemia showed that it was well tolerated and had clinical and biological activity.79 In addition, a phase I study evaluated the combination of the neutralizing anti-BAFF antibody, LY2127399, and Btz. The preliminary results suggested that the safety profile is similar to Btz alone. An ORR of 55 % was noted despite prior Btz therapy in 65 % of patients.80
Antibodies Targeting Bone Effects Of Myeloma
Tumor cells and stromal cells interact via adhesion molecules and cytokine networks to simultaneously promote tumor cell survival, drug resistance, angiogenesis, and disordered bone metabolism. Antibodies targeted against the mediators of these interactions especially retard development of end-organ complications. To date, these efforts have been restricted to the suppression of myeloma-related bone disease.
Receptor Activator of Nuclear Factor-kappa B Ligand
Rank ligand (RANKL) is an essential mediator of osteoclast function, formation, and survival. Increased expression of RANKL promotes bone loss in osteoporosis and contributes to the development of bone lesions in MM. Preclinical studies demonstrated that inhibition of RANKL led to reduced serum paraprotein levels and tumor burden in mouse myeloma models.
Denosumab (Dmab) is a fully human anti-RANKL MoAb. It has demonstrated some efficacy in patients with plateau-phase or relapsed MM, as evidenced by suppression of the bone turnover marker serum C-terminal telopeptide of type 1 collagen (CTX). However, no patient achieved the protocoldefined objective response criteria of CR or PR. Eleven (21 %) subjects with relapsed MM and 19 (46 %) subjects with plateau-phase MM maintained SD for 16.5 months and 18.3 months, respectively.81
A phase III, randomized, double-blind, active controlled trial compared Dmab with zoledronic acid (ZA) for delaying or preventing skeletal-related events (SRE) in patients with advanced cancer and bone metastases (excluding breast and prostate) or MM. Dmab was found to be noninferior to ZA in delaying time to first on-study SRE (hazard ratio [HR], 0.84; p=0.0007). Although directionally favorable, Dmab was not statistically superior to ZA in delaying time to first on-study SRE or time to first-andsubsequent SRE. OS and disease progression were similar between both groups. Osteonecrosis of the jaw occurred at similarly low rates in both groups. Hypocalcemia occurred more frequently with Dmab, while acutephase reactions after the first dose and renal adverse events occurred more frequently with ZA.82 Raje et al. did a subset analysis of patients with MM. Of the 1,776 patients randomized, 10 % had MM (93 in the ZA arm; 87 in the Dmab arm). The OS favored ZA over Dmab (HR, 2.26; 1-year OS 97 % versus 83 %). However, the outcomes are difficult to interpret due to the small sample size and imbalances in baseline disease characteristics, stem cell transplant therapy, and consent withdrawal or loss of follow-up.83 A randomized double blind phase III trial comparing Dmab with ZA for the treatment of bone disease in newly diagnosed MM is currently underway.84
Dickkopf-1
Dickkopf-1 (DKK1), a soluble inhibitor of Wnt signaling secreted by myeloma cells contributes to osteolytic bone disease by inhibiting the differentiation of osteoblasts. High serum DKK1 levels were correlated with focal bone lesions in MM.85
BHQ880 is a fully human anti-DKK1 MoAb. Although BHQ880 had no direct effect on myeloma cell growth, BHQ880 increased osteoblast differentiation, neutralized the negative effect of myeloma cells on osteoblastogenesis, and reduced IL-6 secretion.86 Furthermore, in a SCID-hu murine model ofhuman MM, BHQ880 treatment led to a significant increase in osteoblast number, serum human osteocalcin level, and trabecular bone.87
A phase IB multicentric, dose-determination study of BHQ880 in combination with anti-myeloma therapy and ZA in patients with RRMM and prior SRE was conducted. It revealed that the combination was well tolerated with a general trend towards increased bone mineral density.88
Activin A
Activin A is a transforming growth factor-beta (TGF-β) superfamily member most commonly associated with embryogenesis and gonadal hormone signaling.89 In addition, activin A is also involved in bone remodeling by stimulating osteoclasts growth and inhibiting osteoblasts differentiation.90
RAP-011 is a chimeric antibody, derived from the fusion of the extracellular domain of activin receptor IIA and the constant domain of the murine IgG2a. ACE-011, a protein therapeutic based on the activin receptor IIA, is a novel bone anabolic agent. A phase I study using a single dose of ACE-011 was conducted in postmenopausal women. ACE-011 caused a rapid, sustained, and dosedependent increase in serum levels of bone-specific alkaline phosphatase (BSAP), a marker of bone formation, while decreasing CTX, a marker of bone resorption. In MM a phase II trial is ongoing. Patients are treated with Mel, Thal, and prednisone, and randomized to receive either monthly doses of ACE-011 or placebo for up to 3 months. Preliminary results showed clinically significant increases in biomarkers of bone formation, improvement in skeletal metastases, and decreases in bone pain as well as antitumor activity.91
Conclusions
MM exhibits a number of potentially valuable targets for MoAb therapy. Daratumumab has shown encouraging results as monotherapy in patients with heavily pretreated and refractory MM. The combination of Elo with Len/Dex, and daratumumab with Btz/Len also seem to be promising. Results from other preclinical studies and clinical trials in RRMM have suggested that MoAbs are likely to act synergistically and overcome resistance of various traditional therapies (Dex), immune modulators (Thal, Len), and cytotoxic therapies (proteasome inhibitor Btz).
At present, it remains to be defined how MoAb therapy can most productively be incorporated into the current therapeutic paradigms that will help to achieve longer disease control and significant survival gains in patients with MM either as short-term induction therapy, frontline treatment in transplant ineligible patients, or long-term maintenance therapy post-ASCT/induction cytotoxic therapy.
Future experiments may be directed at inclusion of MoAbs in standard therapy against MM, evaluation of targets, the antibodies against which will hinder the progression from smoldering to symptomatic MM, and of biomarkers to identify patient subsets that are most likely or unlikely to respond to specific MoAbs.