
Evolution of Knowledge in Essential Thrombocythaemia Disease
Presented by: Gunnar Birgegård
Division of Medical Sciences, Haematology, Uppsala University, Uppsala, Sweden
Essential thrombocythaemia (ET) is a serious disease with significant morbidity and mortality and can sometimes transform to acute myeloid leukaemia (AML).1–3 ET must be differentiated from other myeloproliferative conditions such as polycythemia vera (PV) and early primary myelofibrosis (PMF 0/1) because management of the conditions may differ. The World Health Organization (WHO) 2008 criteria state that to differentiate ET from PMF 0/1 it is necessary to perform a bone marrow biopsy.4 The importance of this guidance was emphasised by a study in which bone marrow from patients (n=1,104) who had all previously been diagnosed with ET were reexamined by representatives from seven centres of excellence.5 True ET was confirmed in 891 (81%) cases but the diagnosis was revised to PMF 0/1 in 180 cases (16%) and 33 (3%) were non-evaluable. This study also showed that the 15-year cumulative incidence of AML was higher with PMF 0/1 than ET (11.7% versus 2.1%, respectively) and similarly for myelofibrosis (16.9% vs. 9.3%). In ET diagnosis, therefore, bone marrow samples must be scrutinised by a pathologist who is familiar with the current criteria, and cardiovascular risk factors should be assessed.9,10
DNA sequencing studies indicate that in ET, 50–60% of patients have a mutation in the Janus kinase gene (JAK2
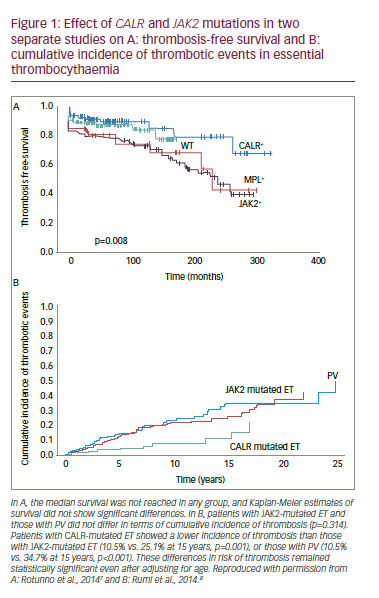
) and 5–10% have a mutation in the thrombopoietin receptor gene (MPL).6 A further 20% of ET patients have a mutation in the calreticulin (CALR) gene. This provides a genetic diagnosis in over 85% of ET patients. Identifying these mutations is important because patients with CALR mutations have improved thrombosis-free survival and decreased incidence of thrombotic events than those with JAK2
mutations (Figure 1).7,8 These findings raise questions over whether JAK2
mutated ET patients have more thrombosis than JAK2
negative patients and whether genetic screening should be included in risk score models. One study (n=891) showed that JAK2
was influential among the factors putting patients at high risk of thrombosis (HR: 2.04) in addition to age >60 years (HR: 1.50) and cardiovascular risk factors (HR: 1.56).9 Another study, however, showed that a similar thrombosis risk stratification was produced when using only cardiovascular risk factors and previous thrombosis.10 It may therefore be necessary to devise individually tailored thrombosis risk assessments in ET that can include mutation screening where it is considered appropriate.
In ET, the entire intra-vessel milieu is prothrombotically enhanced; various cell types are activated, including white blood cells, platelets and endothelial cells. This leads to progressive occlusion and thrombosis of blood vessels up to medium-sized arteries. Inherited prothrombotic factors also increase risk.11–13 For many patients with low risk disease and no major contraindications, low dose (40-100 mg) aspirin is recommended for managing ET.14,15 Another important treatment approach in high-risk ET is cytoreduction using hydoxyurea (HU) or anagrelide. The Primary Thrombocythaemia (PT1, n=776), ANAgrelide compared with HYdroxyurea in WHO-classified Essential Thrombocythemia (ANAHYDRET, n=259) and the Evaluation of Xagrid
Efficacy and Long-term Safety (EXELS, n=3,643) studies have shown that cytoreduction therapy reduces thrombosis in high-risk ET patients and provides similar low rates to that of platelet reduction.16–22
Despite advances in treatment, there are substantial unmet needs in ET. These include a marker of biological activity and advanced disease, drugs that address the underlying disease and targets for such drugs need to be identified. In addition, the implication of mutation ‘triplenegative’ ET patients (i.e. JAK2-, CALR- and MPL-negative) needs to be determined to establish the disease mechanisms and appropriate treatment in individuals with that genetic profile.
Case example
An unusual case of essential thrombocythaemia(Presented by Dr T Lado, Cives Servicio de Hematología Clinica, Hospital Clínico Universitario de Valladolid, Spain)
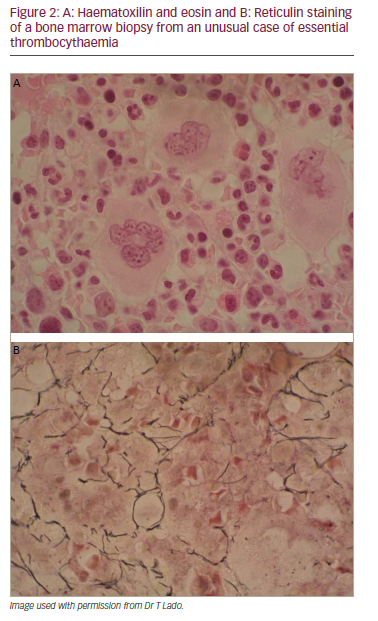
A 34-year-old woman with painful splenomegaly showed normocellular bone marrow with prominent megakaryocytes, no clonal cells and normal cytogenetics. Following splenectomy, histopathology showed congestion. One week later she was readmitted with headache and abdominal pain. She also had a burning sensation in the palms and soles and facial blushing. Laboratory findings showed Hb 11.3 g/dL, leucocytes 23.58 x 109/L, neutrophils 18.79 x 109/L, platelet count 1776 x 109/L, GGT 1,16 U/L and alkaline phosphatase 208 U/L. A CT scan showed widespread portal thrombosis of main veins and spleen vein and partial thrombosis of the superior mesenteric vein. The cerebral CT scan was normal. The diagnosis was severe reactive thrombocytosis secondary to splenectomy with possible transient ischaemic attack (TIA). Anticoagulant therapy and low-dose aspirin was started. One month later she had a platelet count of 3,244 x 109/Land series 5 thrombapheresis treatments were performed. In a revised haematological approach, bone marrow aspiration showed megakaryocytic hyperplasia; bone marrow biopsy revealed increased megakaryocytes and moderate reticulin fibrosis compatible with ET (Figure 2). The patient was Philadelphia-negative but in peripheral blood, 23% of alleles were JAK2 mutated. This case was diagnosed as ET according to the proposed revisions to the WHO ET criteria and was considered suitable for first-line therapy for high-risk patients and given hydroxyurea with low-dose aspirin. Platelet counts were restored to within the normal range by 12 days and were subsequently maintained at that level.
Data from case reports are not representative of the general population with ET and therefore cannot be extrapolated into general clinical practice.
This case showed that the onset of ET without thrombocytosis is a diagnostic challenge. It emphasises that
• indolent lymphomas and myeloproliferative neoplasms (MPNs) are the most prevalent haematological malignancies associated with splenomegaly;23,24
• in unexplained splenomegaly, even where the haemogram is normal, it is important to suspect MPN and screen for JAK2 mutation;25,26
• up to 30% of ET cases have splenomegaly at diagnosis.27,28
The case indicates that thrombapheresis is not always appropriate but its use should be considered on an individual basis. It is clear, however, that all patients with suspected ET should be carefully evaluated by a pathologist who is well acquainted with the new WHO criteria.29
How to Identify and Manage Cutaneous Manifestations Associated with Myeloproliferative Neoplasms
Presented by: Selim Aractingi
Department of Dermatology, Cochin Hospital, Paris, France
The skin can be regarded as an early diagnostic tool for recognition of patients with MPNs. It is a large barrier organ at the interface between the exterior and the dermatological surface and is composed of many different cell types, changes in which can reflect disorders within various different body systems. MPNs are responsible for various signs in the skin, mainly related to microvascular disorders.30
In ET, pruritus is an important sign that can aid diagnosis. Pruritus results from mast cell modification and secretion of various inflammatory mediators including histamines, 5-hydroxytryptamine, acetylcholine, substance P, leukotrienes, bradykinin, proteases, interleukins 2, 4, 6 and 31 and others. These stimulate afferent nerve fibres triggering the urge to scratch.31,32 Pruritus is found in 5–69% of PV cases, 3–46% of ET cases and 16–54% of myelofibrosis cases.33–36 Pruritus can occur as two types: the classic form or the type that is induced by water contact (aquagenic pruritus). The latter is found in 30–50% of patients with PV and 30–50% of patients with ET.33,35 It is important that physicians look for pruritus and investigate its cause when it is present.
In ET, microvascular lesions manifest as various types of skin conditions including erythromelalgia (in 6% of patients, often on palms and soles) and spontaneous ulcers (in 4–5%), livedo reticularis (in 3%, usually on feet and legs) and superficial phlebitis (in 8.6%, resulting from occlusion of larger vessels). Rarer symptoms include acrocyanosis, Raynaud’s phenomenon, blue toes, digital gangrene and vasculitis.30,37 Other types of lesions include neutrophilic dermatosis and vasculitis. These symptoms are therefore highly important in identifying patients with early MPNs or for indicating where treatment adaption is needed in previously diagnosed patients.
Case example
Treatment-induced skin ulcer in a patient with essential thrombocythaemia (Presented by Dr C Nakaseko, Department of Hematology, Chiba University Hospital Chiba, Japan)
Thrombocytosis (900 x 109/L) was detected in a 59-year-old Japanese male during a physical examination. ET was subsequently diagnosed after tests that included bone marrow aspiration. Thrombosis risk was considered low and treatment was initiated with lowdose aspirin. After one year, the platelet count increased to 1,790 x 109/L. Hydroxyurea (HU) 500 mg/day was started but failed to decrease the platelet count below 1,100 x 109/L. Consequently, the HU dose was increased to 1,000 mg/day. Three years later the platelet count increased to 1,580 x 109/L and HU dose increased to 1,500 mg/day after which the platelet count remained at 800 x 109/L. One year later the patient had a small cut on the right heel with ulceration (Figure 3). HU was reduced to 1,000 mg/day and silver sulfadiazine cream was started. The skin ulcer showed only slight improvement and remained severe and the platelet count increased to >2,000 x 109/L without any bleeding tendency and HU dose was not increased. Two years later, when anagrelide was approved in Japan, 1 mg/day b.i.d. of this drug was started with no apparent adverse reactions and the platelet count decreased from 2,300 x 109/L to 620 x109/L. The dose of anagrelide was then increased to 1.5 mg/day b.i.d. and HU was reduced and then discontinued. The skin ulcer healing improved and later completely resolved. The platelet count was subsequently maintained in the normal range.
This case showed that an intractable skin ulcer developed after increasing the dose of hydroxyurea given to treat an uncontrolled
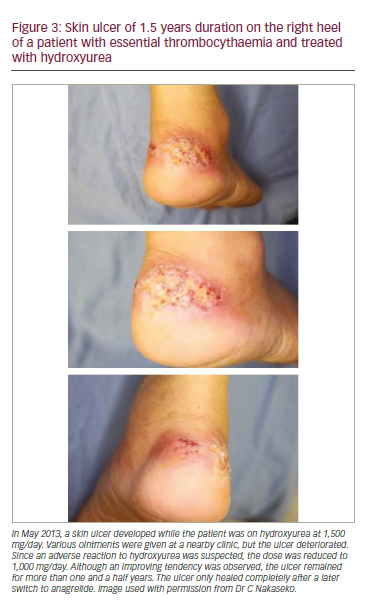
platelet count in ET. Treatment with anagrelide provided successful platelet count control and enabled the discontinuation of HU and consequent resolution of the skin ulcer. Anagrelide is therefore a valid option in cases of MPN with adverse reactions to hydroxyurea.
The possible reason why HU-induced skin ulcers, such as this case, occur more commonly in the lower limbs may be a result of greater venous pressure at these locations which is a known causal factor of ulceration.38 Other risk factors for these ulcers are increased age and female sex, but the evidence for this is weak.39,40 Experience shows that effective management of pruritus in ET requires continuous collaboration between haematologists and dermatologists.
Identifying Key Aspects in Women’s Health When Managing Essential Thrombocythaemia
Presented by: Claire Harrison
Guy’s and St Thomas’ NHS, London, UK
Women of all ages are affected by MPNs, and the influences and effects of contraception, pregnancy and menopause are important considerations. Women with PV are more likely to have venous thrombotic events and microcirculatory disturbances, but tend to be older at diagnosis, as shown in the EffiCacy and safety of Low-dose Aspirin in Polycythemia vera (ECLAP) study.41 Splanchnic venous thrombosis was shown to occur most frequently in young women, and five in every six cases of Budd-Chiari syndrome (a common

consequence of MPNs) were female. Men, however, were more likely to have peripheral arterial thrombotic events and myocardial infarction.41 In another large study, among 19 cases of abdominal vein thrombosis, 17 were female and, in a further large study of MPNs, seven of nine patients with cerebral venous thrombosis were female.42,43
A British Journal of Haematology guideline, developed in the UK for investigation and management of adults and children with thrombocytosis, states that the combined oral contraceptive pill (oestrogen-based) should be discouraged in patients with MPN.44 In addition, ovarian stimulation therapy is associated with a thrombosis risk; individual cases should be assessed and thromboprophylaxis offered where appropriate. Furthermore, hormone replacement therapy may slightly increase the risk of venous thromboembolism (VTE) and should only be used in patients without additional risk factors and no thrombosis history. Key issues to discuss with MPN patients concerning pregnancy are risks of poor pregnancy outcome, thrombohaemorrhagic events and the teratogenicity of drug treatments.45
A meta-analysis of 461 pregnancies in women with ET (n=461) showed a 50–70% live birth rate.46 This analysis also captured first trimester loss (25–40%), late loss (10%), placental abruption (3.6%), intrauterine growth retardation (IUGR, 4.5%) post-partum thrombosis (5.2%) and ante/postpartum haemorrhage (5.2%). There is generally less information available on the effects of PV in pregnancy but outcomes appear worse than with ET. A case series of 18 pregnancies and 20 single case reports of PV show a similar live birth rate to ET (60%), first trimester loss (21%), late loss (15%), IUGR (15%) premature delivery (13%).47 Among these there were two cases of pulmonary embolism, one post-partum haemorrhage and four cases of pre-eclampsia. In addition, there was one death involving deep vein thrombosis and other thrombotic events. There is also very little information on PMF in pregnancy. A series of four cases and four single reports showed a 50% risk of foetal loss but no maternal thrombosis or haemorrhage.48
Predicting obstetric complications in ET is problematic. Decreases in platelet counts may correlate with improved outcome.49 However, disease complications and past pregnancy outcomes may not predict subsequent events and outcomes.50 In pregnant women with MPN, measuring the uterine artery Doppler to assess pulsatility index at 20–24 weeks is useful for predicting outcome or indicating a need to change therapy.51
Various factors put pregnant women with MPN at high risk of thrombosis, including previous venous or arterial thrombosis, previous haemorrhage (due to MPN) elevated platelet count, diabetes and hypertension (see Table 1). In addition, MPN may have caused complications and poor outcomes in previous pregnancies, including multiple miscarriage, stillbirth, IUGR and severe eclampsia.45
Recommended management of MPNs in pregnancy includes a pre-conceptual meeting, determination of thrombosis risk in each woman and, where necessary, control of the platelet count.45 It is also necessary to control the haematocrit to within the gestation-specific range and to use low molecular weight heparin post-partum to reduce the thrombosis risk.44
Case example
A 39-year-old woman with thrombocytosis and a history of: spontaneous pneumothorax (six years), high platelet count (six years), endometriosis (laparoscopy-oophrorectomy and partial right oophorectomy [occurring 12 and 14 years earlier, respectively]) and episodes of amausis fugax. Two years previously, she had lower extremity oedema and hepatomegaly; a liver biopsy indicated sharpened Budd-Chiari syndrome. She was JAK2– and thrombophilia test-negative. A diagnosis of MPN was rejected; the thrombosis was considered secondary to risk factors (oral contraceptives and smoking). Contraceptives (taken for 10 years) were stopped and treatment with acenocumarol was started.
The patient had a platelet count of 942 x 109/L and was found to be CALR exon 9 positive (type 1 mutation). A bone marrow biopsy showed increased numbers of enlarged mature megakaryocytes and mild to moderate fibrosis on reticulin staining. These findings were compatible with a revised diagnosis of CALR-positive ET with high thrombosis risk.
This case highlighted some issues in the treatment of young ET patients, e.g. whether analysis of CALR mutations should be routinely performed. The usefulness of CALR mutations in MPNs, particularly in Budd-Chiari syndrome, is unclear. Some investigators suggest performing CALR analysis only in JAK2-negative cases, whereas others would never perform such analyses.52–56 The test is quite simple and allows a correct diagnosis, although the reported incidence of CALR in MPN is low, no higher than 2.3%.54 There is, therefore, a lack of consensus on this genetic testing that needs further debate and investigation. The case also emphasises the need to perform a bone marrow biopsy at an early stage, which would assist the diagnosis and should be done prior to genetic testing. This patient is clearly unsuitable for oestrogen-based combination contraception) but progesterone-based, barrier, IUDs or long-term methods would be more suitable. In patients with MPN and additional risk factors for VTE, it may be inappropriate to prescribe hormone replacement therapy since this could also increase the thrombosis risk.44
Management of Elderly Patients with Essential Thrombocythaemia
Presented by: Jean-Jacques Kiladjian
Clinical Investigations Centre, Hôpital Saint-Louis, Paris, France
Age is a marked risk factor in MPN; 75% of patients are diagnosed >60 years and 51% are over 70 years.9,57,58 Despite this, there is a lack of data on MPN in elderly patients. The EXELS registry study provided a unique opportunity to investigate this group because among the 3,598 patients recruited in 13 European countries, 395 were >80 years at registration.22 This was the largest elderly ET patient cohort studied to date. In patients <65 years, 65–<75 years and ≥75 years, the proportions receiving anagrelide were 72%, 17% and 12%, respectively, whereas the proportions receiving other cytoreduction therapy were 34%, 34% and 32%, respectively.59 This difference may reflect the current recommendation to use HU in younger patients with caution and a possible concern that the incidence of serious side effects due to agagrelide treatment is higher in patients aged 60 years or older than in those under 60 years.60
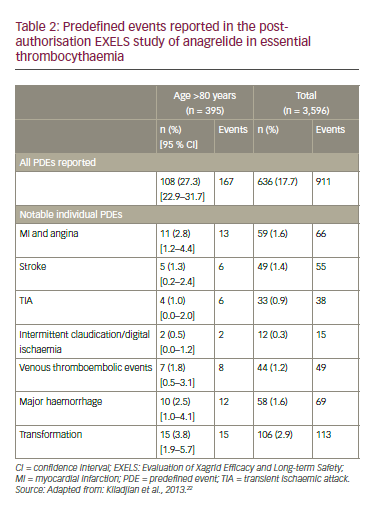
In the EXELS study, the incidence of thromboembolic events was slightly higher in patients aged >80 years than in the whole study population (6.3% versus 5.2%, respectively).22 The risks of stroke (1.3% versus 1.4%), TIA (1.0% versus 0.9%) and venous thrombotic events (1.8% versus 1.2%) were similar in both groups (Table 2). The study showed that the management of elderly ET patients is homogeneous across European countries and that the characteristics of elderly ET patients do not substantially differ from the whole ET population. Interestingly, the haematological response to cytoreductive therapy was similar in different age groups; the elderly showed no greater toxicity. In addition, no new safety concerns regarding treatment in elderly patients were observed.22
Case example
Essential thrombocythaemia in an elderly patient
(Dr S Alimam, Department of Haematology Guy’s and St Thomas’ NHS Trust, London UK)
An 84-year-old female smoker with >40 year pack history was found on routine investigations to have a platelet count of 1,157 x 109/L. ET was suspected and she was found to be CALR-positive with no splenomegaly. She had a myocardial infarction (MI) 20 years earlier with ongoing hypertension and high cholesterol. The patient was started on aspirin 75 mg/day with optimised hypertension and cholesterol control and smoking cessation. She was given HU 500 mg/day, which was well tolerated but produced no change in platelet count. Increasing the HU dose to 1,000 mg/ day on weekdays and 500 mg/day at weekends provided good platelet control but she was increasingly intolerant with ongoing thinning hair, nausea, weight loss and anaemia (Hb<100 g/L). She had fatigue, oral ulcers and feared suffering a stroke. The patient then opted for a treatment break, which improved the symptoms but the platelet count rapidly doubled. Further options including a higher platelet threshold or interferon injections were rejected. Anagrelide was inappropriate due to her cardiovascular history. The patient accepted pulsed busulfan 4mg/day for 14 days which was repeated if platelets >400 x 109/L after 28 days. The patient appeared to tolerate the treatment well; the pulsed dosing with time off treatment was appreciated and platelet counts were reduced to <500 x 109/L (Figure 4).
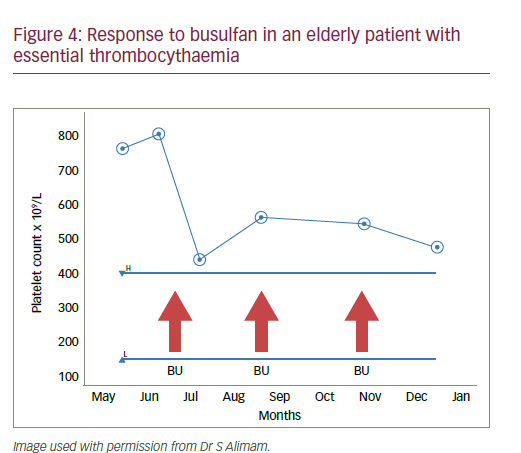
This case stresses that increased cardiovascular risk factors and cardiovascular events are real complications in elderly patients with ET and that higher rates of therapy-related side effects are more likely in this age group. This experience shows that treatment must be individualised and it is necessary to balance side effects, quality of life (QoL) and the target platelet count. In addition to busulfan, other drug treatments and drug combinations could also be considered in elderly ET patients. This case also shows that treatments must be suspended and changed when side effects are intolerable. It may also be possible to accept less strict control platelet counts in order to reduce side effects such as mucocutaneous lesions which impair QoL in the elderly.
Identification and Management of Cardiovascular Risk Targets in Essential Thrombocythaemia Conclusion
Presented by: Manuel Martinez-Sellés
Department of Cardiology, Gregorio Marañon Hospital, Madrid, Spain
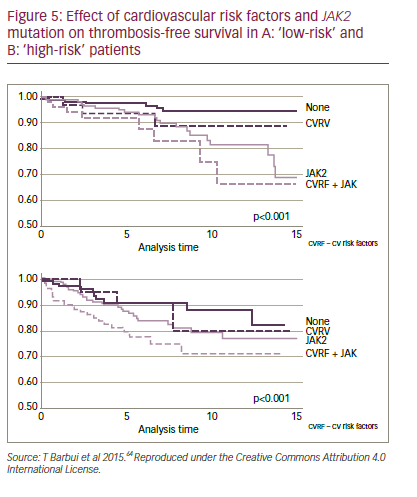
In all individuals, especially those with ET, it is essential to reduce cardiovascular (CV) risk factors for thrombosis mainly through avoiding tobacco, increasing activity and opting for good nutrition (to achieve blood glucose <110 mg/dL, blood pressure <140/90 mmHg, cholesterol <190 mg/dL, LDL <115 mg/dL and body mass index [BMI]<25 kg/m2).61–63 The classical ET score systems included only age (>60 years) and thrombosis history.9 However, newer systems, such as the International Prognostic Score for Essential Thrombocytopenia (IPSET-Thrombosis) model, assess more parameters with differing risk levels (points) assigned to them.9 These include CV risk factors (one point) thrombosis history (two points), age >60 years (one point) and JAK2-positive (two points). A study by Lekovic et al.10 produced a slightly different score system in which one CV risk factor (one point), >one CV risk factors (two points) and thrombosis history (two points) were the key parameters. In this model, a one- to two-point score has a thrombosis risk of 16.7% but a score of more than two points has a risk of 60%. In these systems, CV risk factors are as important as age. Patients with any CV risk factors should not be considered ‘low risk’, especially when the patient is JAK2 positive. It has recently been proposed that ‘low risk’ patients should be subdivided into ‘very low risk’ and ‘low risk’ depending on the presence of the JAK2 mutation (Figure 5).64 These classifications are important, since they inform treatment strategies.
In all ET patients, CV factors should be assessed and treated where present. Subsequently, if CV history and ECG are normal, cytoreductive therapy can be started, if they are not normal, cardiac evaluation should be initiated and include echocardiography and a stress test. Treatment that addresses these factors substantially improves the prognosis.65
ET patients over 60 years, therefore, should be given cytoreductive therapy, but for younger patients treatment decisions should be based on risk factors.66,67 The most important predictor of thrombosis is the presence of CV risk factors and previous thrombosis.10
Another important risk factor is obesity, but reducing and maintaining low body weight is challenging and few patients achieve it, so other approaches to managing ET need to be taken. In ET, the pathogenesis is insufficiently understood but the most likely mechanism is atherosclerosis, which is more likely in obese patients and CV risk factors which can lead to coronary thrombosis resulting from plaque rupture.10, 68–70
Conclusion
In recent decades, considerable progress has been made in identifying and managing ET and reducing the risks associated with it.71 Despite this, substantial unmet needs remain which include better markers for biological activity and advanced disease, improved understanding of the pathogenic mechanism and more effective and safe/tolerable treatments.72 It is not clear which is the best prognostic score system for ET. The traditional risk factors are age (≥65 years), previous thrombosis and CV risk factors.9,10 In clinical studies it may be possible to improve the prognosis by monitoring additional factors but it is not clear which of them to use. Some factors such high BMI (>29.3 kg/m2),low physical activity and adult-onset diabetes increase thrombosis risk in ET.73 In addition, the presence of JAK2 and, to a lesser extent, CALR mutations also increase this risk74 but it is not possible to conduct a study that could properly evaluate all of these factors.
In the treatment of women with ET, cytoreduction therapy is affected by contraception, pregnancy and menopause and should be altered accordingly.44–46,60 This includes stopping or altering oestrogen-based contraception and hormone replacement therapy should be altered to minimise thrombosis risk.44,45
The effects of ET often manifest in the skin and physicians should be aware of these symptoms, as both diagnostic signs and as issues in drug therapy.30,37 There is no clear consensus on whether tests for ET should be performed routinely in patients with pruritus but an early bone marrow biopsy provides a good indication and would justify genetic testing for JAK2, MPL and CALR mutations.4,5,7,8,29 In the future, increased awareness and willingness to perform these tests and the development and wider use of effective treatments in ET have the potential to substantially improve outcomes in this common and serious haematological disease.











