
Hereditary hyperferritinemia-cataract syndrome (HHCS) is a rare autosomal dominant disorder associated with high plasma ferritin concentration without iron overload and early-onset bilateral cataract. HHCS was first described in 1995,1–3 and since then over 30 mutations, mainly point mutations and deletions, associated with HHCS have been reported.4–6
Isolated hyperferritinemia with plasma ferritin concentrations above 300 μg/l in men and 200 μg/l in women in an asymptomatic patient and in absence of inflammation and liver disease, most likely indicates increased iron stores and requires further investigation whether or not it is accompanied by iron overload – in order to differentiate between hereditary haemochromatosis (HH), haemosiderosis, ferroportin disease, aceruloplasminemia, atransferrinemia and HHCS.7
Under normal physiological conditions, plasma ferritin reflects the amount of iron stored in tissue macrophages – apart from infectious, inflammatory, autoimmune and malignant diseases, where plasma ferritin also acts as an acute-phase reactant. Ferritin is composed of different subunits, L, H and G, which arrange to form different isoferritins. The synthesis of these ferritin subunits is regulated by iron availability through an iron regulatory system. Iron-responsive elements (IREs) are one component of this system and are mRNA stemloop structures located in the 5′ or in the 3′ untranslated regions (UTRs) of the genes involved in iron metabolism. IREs are crucial in regulating the cellular iron metabolism and coordinating the synthesis of various proteins involved in the iron metabolism, including transferrin receptor for cellular iron uptake, ferritin for intracellular iron storage and erythroid-specific δ-aminolevulinate synthase (δ-ALAS) for iron use for haemoglobin synthesis. The stem structure of the mRNA is interrupted by several bulges with a terminal loop that is essential for binding of cytoplasmic proteins called iron regulatory proteins (IRPs). IRPs are the other components of the iron regulatory system, which bind to IREs in response to iron deficiency, induce expression of transferrin receptors (increasing iron uptake) and repress the synthesis of ferritin and δ-ALAS (decreasing iron storage and iron use). The opposite occurs in conditions of iron excess.6,8–10
The deregulation of ferritin production in HHCS is caused by mutations in the IREs of the FTL gene – interfering with the high-affinity interaction between IREs and IRPs, leading to disrupted down-regulation/negative regulatory control by IRPs of L-ferritin mRNA translation and resulting

in excessive production of L-ferritin regardless of the iron level. The only known organ damage in HHCS is cataract, which appears to be a consequence of accumulation of L-ferritin within the lens fibres.8–10 Thus, the only treatment available for HHCS is cataract surgery.
Case History
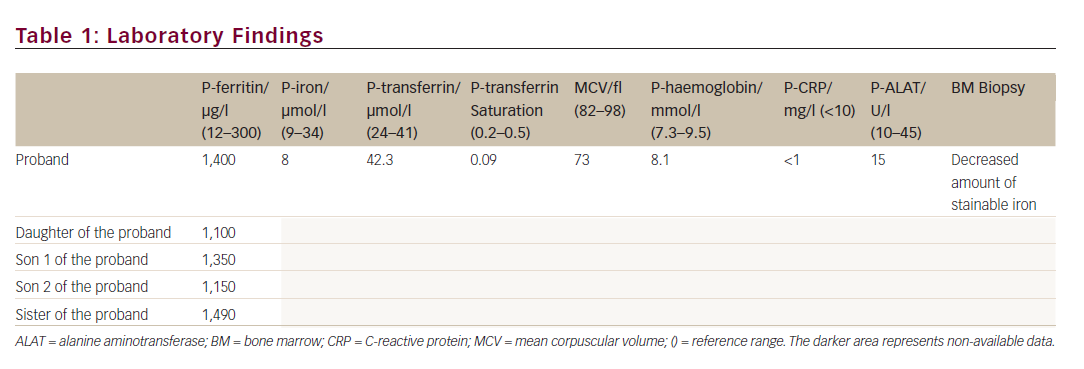
A 44-year-old Caucasian woman was suspected of having HH in 2011, when she was referred from the medical department with a plasma ferritin of 1,400 μg/l. She was asymptomatic, and the physical examination was normal. Blood samples showed low plasma iron, low transferrin saturation, decreased mean corpuscular volume (MCV), normal inflammatory markers and normal haemoglobin (see Table 1). The patient was negative for HFE gene mutation (related to HH), had normal liver cell counts and liver size and the bone marrow displayed low iron deposits. Apart from hyperferritinemia, the only abnormality was a medical record showing bilateral cataract surgery performed 21 years ago. Thus, the combination of hyperferritinemia not due to iron overload and previous bilateral cataract surgery led the clinicians on the track of HHCS.
Methods
Plasma ferritin levels and genomic DNA were extracted from peripheral whole blood of each subject.
Molecular Diagnosis
Genomic DNA was purified from leukocytes using the QIAamp DNA Blood Mini QIAcube Kit. The 5′ UTR region of the FTL gene was polymerase chain reaction (PCR)-amplified using forward primer 5′-CTATGTGCTCCGGATTGGT-3′ and reverse primer 5′-AGAGAGGTAGGTGTAGGAGG-3′. PlatinumTaq DNA Polymerase was used for PCR amplification. The following conditions were used: 120 seconds at 95°C, followed by 30 cycles of 30 seconds at 95°C, 30 seconds at 60°C and 40 seconds at 72°C followed by a final 7 minutes at 72°C. PCR products were purified using QIAquick PCR purification kit. Sequencing was performed using BigDye Terminator v3.1 Cycle Sequencing Kit and subsequently purified using BigDye XTerminator purification kit. Capillary electrophoresis was performed on an ABI 3,500 genetic analyser. All kits were used according to manufacturer’s instructions.
Results
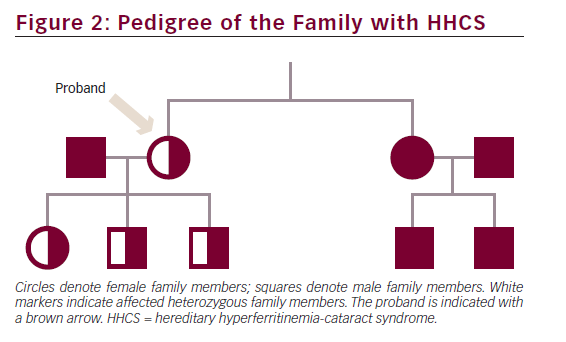
The proband, her three children and her sister were heterozygous for a point mutation in the IRE located in the 5′ UTR of the FTL gene on chromosome 19q: G32T (see Figure 1), previously reported as the Paris 2 mutation by Martin et al.11 Data on the consanguineous parents of the proband were not available. The two children of the sister were both negative for the point mutation (see Figure 2).
Discussion
When facing a high plasma ferritin concentration in an apparently healthy person, it is important – besides excluding HH in order to prevent organ damage caused by iron overload – to take HHCS into account, despite its rarity, because a mistaken diagnosis of HH could lead to rapid development of phlebotomy-/iron chelation therapy-induced irondeficient, microcytic anaemia and unnecessary liver biopsies. Thus, the differential diagnosis between HH and HHCS is essential and prevents unnecessary phlebotomy. In this case, plasma biochemical investigation of iron status, family genomic DNA-screening, and an ophthalmological examination are recommended. Craig et al.12 even propose cataract morphology to be pathognomonic for HHCS. Furthermore, it is important to consider spontaneous de novo mutations in the FTL gene causing non-hereditary hyperferritinemia cataract syndrome (non-H-HCS), which has been reported by Cao et al.13 With this in mind, HHCS should be taken into account even in sporadic cases of early-onset cataract formation.
Patients with HHCS present a versatile phenotype. A clinical variability among subjects sharing the same mutation has been demonstrated, in terms of plasma ferritin levels and time at onset and severity of cataract.9,10,14M/sup> Allerson, Cazzola and Rouault found a close correlation between decreases in the IRPs–IREs binding affinity and clinical severity.
Thus, the severity appears to be determined by the energetics of the binding interaction, and the latter varies among different mutations.15 It is likely that naturally occurring polymorphisms within mRNA regulatory proteins contribute to phenotypic diversity, and that other genetic or environmental modifiers play a role in the severity of the disease. Herein, next-generation sequencing studies could be useful in a prospective setting.











