

Early diagnosis of rare haematological diseases
Rare haematological diseases often present ‘in disguise’, with nonspecific symptoms such as pancytopaenia, decreased circulating levels of one or two cell subsets and splenomegaly. Gaucher disease (GD) provides an excellent paradigm when considering challenges in diagnosing rare haematological diseases. Individuals presenting with GD will only rarely be seen regularly in the general haematology setting, even in areas with prominent Ashkenazi heritage communities. Such rare disorders require a more investigative approach to symptoms on the part of the haematologist. To assist diagnosis in such cases, the doctor should look for those features which do not match with the clinical picture of the usual underlying diseases. For most rare haematological diseases, including GD, sensitive and definitive confirmatory diagnostic tests are available, so confirming the diagnosis is relatively straightforward if the symptoms are recognised early.1 Given the inherent difficulties in the diagnosis of these diseases, the Sherlock Holmes symposia were initiated as an educational initiative to raise awareness of rare haematological diseases. The symposia were designed to aid haematologists to recognise the symptoms of these diseases, to examine the clinical decision making required to support differential diagnoses, to identify appropriate confirmatory testing and to assist in recognising the need for early treatment intervention.
Inspired by the Sherlock Holmes symposia, this article explores the diagnosis of rare haematological disorders by examining a number of informative real-life case studies with a focus on cases involving thrombocytopaenia and/or splenomegaly.
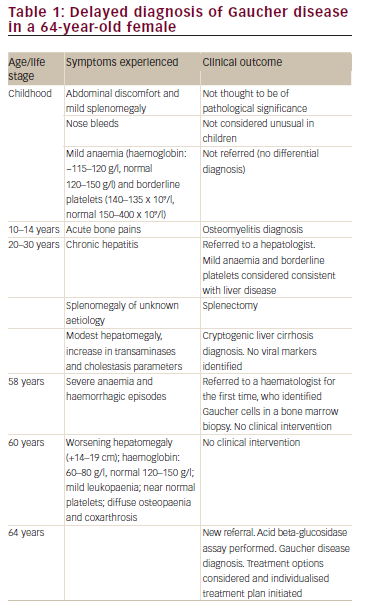
Case study one – delay in diagnosis of rare haematological disorders This case concerns a female with symptomatic GD who was undiagnosed for over 50 years (Table 1). Symptoms suggestive of GD in childhood and adolescence, such as splenomegaly, bone pain and borderline thrombocytopaenia were attributed to other conditions; for example, she received a diagnosis of osteomyelitis for the bone pain. This had a detrimental impact on the patient when she was in her 20s, when she underwent splenectomy due to splenomegaly without any definitive diagnosis, as no histological evaluation of the spleen was available. Given that there is evidence that splenectomy can be detrimental in GD and may result in more aggressive disease in the liver, skeleton and lungs, the splenectomy may have had an ongoing adverse impact on the individual.2 Another clear opportunity for diagnosis was missed in the patient’s late 50s, when she presented with severe anaemia (haemoglobin [Hb] 80 g/l) and haemorrhagic episodes. At that stage, and despite the identification of Gaucher cells in a bone marrow biopsy, diagnosis was still delayed for a further six years. While this is an extreme example, diagnostic delay in GD is common. For example, a review of 136 patients with GD identified a mean diagnostic delay of 10.3 ± 4.1 years from first onset of symptoms.3 The wide range of initial diagnoses in these patients included growing pains, lymphoma, liver disease, osteoporosis, fibromyalgia, collagenosis, cirrhosis, pituitary adenoma and Von Willebrand’s disease; indicating the difficulties facing physicians in formulating an initial diagnosis for GD. Similarly, the failure to recognise the classic symptoms of GD (cytopaenia, hepatosplenomegaly and bone pain) was identified by a global study of 406 haematologists/oncologists, which reported that only 20% considered GD in their differential diagnosis when presented with a patient exhibiting these symptoms.4 Instead, the majority of physicians considered leukaemia, lymphoma and multiple myeloma as the most likely diagnoses.4
Given that there are now effective therapeutic interventions for GD with both enzyme replacement therapies (ERT) and substrate replacement therapies (SRT) being available, it is particularly important that diagnostic delay is avoided and that early opportunities for intervention are not missed.5 This is critical, as treatment can reduce the risk of patients developing bone complications such as osteonecrosis, osteoporosis and lytic lesions, which are irreversible once they are established.1
Splenomegaly
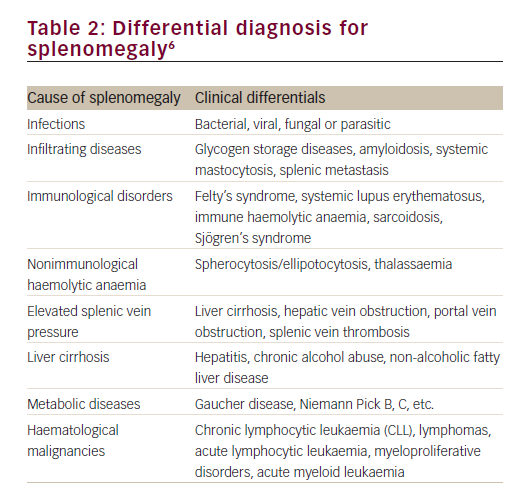
Enlargement of the spleen is a diagnostic clue to a number of conditions of diverse origin including infection, haematological malignancy, red cell disorders, metabolic conditions and other benign infiltrations (Table 2).6 The clinician must integrate signs of splenomegaly with features of hypersplenism in the peripheral blood and other aspects of the clinical history, examination and investigations, in order to make a correct diagnosis of the underlying pathogenic mechanism. The following case identifies the importance of splenomegaly, both in the initial presentation and in the recognition of clinical deterioration due to underlying disease.
Case study two – splenomegaly in Gaucher disease and myelodysplastic syndrome
An active 36-year-old male presented with mild anaemia, thrombocytopaenia and high ferritin levels (1,400 ng/ml, normal
12–300 ng/ml). Two years previously, he was suspected of having genetic haemochromatosis but HFE gene testing discounted this diagnosis. The patient reported lack of energy and tiredness over a period of a few months, which restricted his sporting activities, but experienced no other symptoms except for mild, occasional peripheral sensory loss.
The patient had not received medication for the previous six months. Physical examination showed normal cardiovascular and respiratory function. The liver (4 cm) and spleen (3 cm) were palpable. Neurological examination showed wasting of foot muscles, decreased vibratory sensation below the knees, and loss of pain sensation. Biochemical laboratory tests showed: haemoglobin (Hb) 125 g/l (normal 130–170 g/l); mean corpuscular volume (MCV) 81 femtoliters/cell (normal 80–100 fL); white blood cells (WBC) 6.2 x 109/l (normal 4–10 x 109/l); thrombocytopaenia (platelets 88 x 109/l) (normal 150–400 x 109/l); aspartate aminotransferase (AST) 67 U/l

normal 5–30 U/l); alanine aminotransferase (ALT) 89 U/l (normal 5-30 U/l); gamma-glutamyltransferase (gGT) 35 U/l (6–50 U/l); total bilirubin 1.7 mg/dl (normal 0.3–1.2 mg/dl), unconjugated bilirubin 1.3 mg/dl (normal 0.2–0.7 mg/dl); iron 84 μg/dl (50–175 μg/dl); transferrin saturation 30% (normal 20–50%); ferritin 1400 ng/ml (normal 12–300 ng/ml); hepatitis C virus (HCV) and hepatitis B surface antigen negative; blood film normocytic/microcytic cells; poikilocytosis; glucose-6-phosphate dehydrogenase (G6PD) activity was normal.
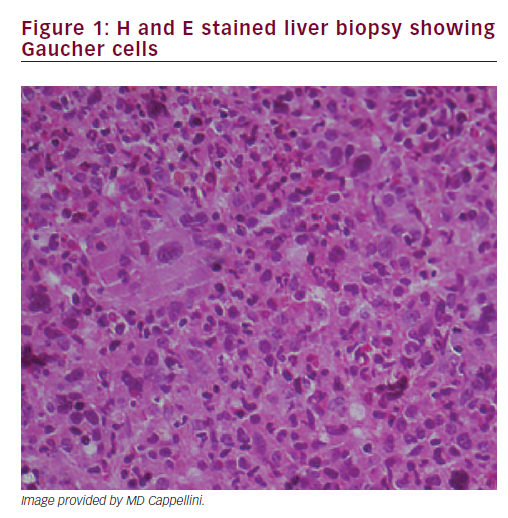
A liver biopsy was performed, which showed Gaucher cells (Figure 1). Diagnosis of GD type 1 was confirmed by glucocerebrosidase activity in leucocytes (2.2 nm/mg/h, normal 11.5 + 3.6). Chitotriosidase activity was 1.48 nm/mg/h (normal 25.75 + 17); genetic analysis: N370S/P401L.
The patient started enzyme replacement therapy (ERT) at 30 U/ kg/2 weeks. After one year of treatment, organomegaly (evaluated by magnetic resonance imaging) and haematological parameters had improved significantly: Hb: 135 g/l (normal 130–170 g/l); MCV 83 fl; WBC 6.7 x 109/l (normal 4–10 x 109/l); neutrophils 5.7; lymphocytes 3.4; platelets 137 x 109/l (normal 150–400 x 109/l); AST 27 U/l (normal 5–30 U/l); ALT 27 U/l (normal 5–30 U/l); gGT 35 U/l (normal 6–50 U/l); total bilirubin: 1.0 mg/dl (normal 0.3–1.2 mg/dl), unconjugated bilirubin: 0.7 mg/dl (normal 0.2–0.7 mg/dl); iron: 118 mg/dl (50–175 μg/dl); transferrin saturation 28% (normal 20–50%); ferritin: 475 ng/ml (normal 12–300 ng/ml) (Table 3).
The patient did well until September 2010, when he complained of marked asthenia, fever and bone pain. Haematological parameters worsened: Hb 103 g/l (normal 130–170 g/l); WBC 23.48 x 109/l (normal 4–10 x 109/l); (neutrophils 85%, lymphocytes 11%, blasts 2%; erythroblasts 3%); platelets 173 x 109/l (normal 150–400 x 109/l); lactate dehydrogenase (LDH) 1115 U/l (normal 135–225 U/l); C-reactive protein (CRP) 0.2 mg/dl (normal <0.50 mg/dl). He had mild hepatomegaly and an enlarged spleen (17.3, 5 cm below the costal margin). Due to increased leukocytes, high LDH and blasts in peripheral blood and reappearance of anaemia, a bone marrow aspirate and bone marrow biopsy were performed.
An ‘unclassifiable myelodysplastic syndrome’ was diagnosed with an unfavourable karyotype: 46XY, del (3)(q21), del (20)(q12) according
to World Health Organization (WHO 2008) classification system.7 The patient started hydroxyurea (500 mg) with blood transfusion support while continuing ERT at the same dosage. During the next few months he had repeated infections. Haematological status remained stable until July 2011, when azacitidine was initiated in response to a significant increase in leukocytes (54 x 109/l, normal 4–10 x 109/l) with blast cells in peripheral blood and platelets increased (691 x 109/l, normal 150–400 x 109/l).8 The patient was admitted to hospital for investigation. A bone marrow biopsy showed a significant increase in blast cells suggesting evolution to overt acute myeloid leukaemia (AML). He was treated with induction chemotherapy and achieved clinical remission. However, despite the use of intensive consolidation strategies with high-dose cytarabine, he relapsed and died due to the secondary AML.
Thrombocytopaenia
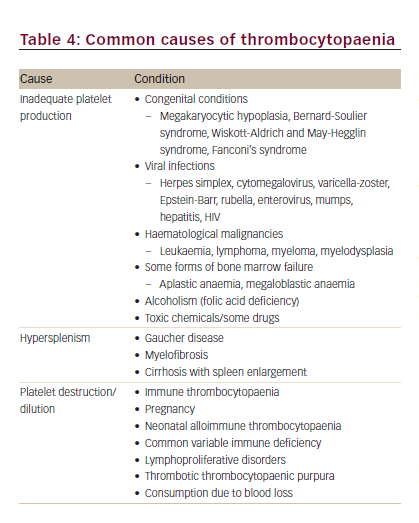
A reduced platelet count is a diagnostic clue to a number of conditions of diverse origin such as rare congenital and, in particular, acquired thrombocytopaenias, infection, haematological malignancy, immunological disorders, viral infections and metabolic disorders (Table 4).9 Diagnosis based on the clinical presentation and history should start with an evaluation of all blood counts and confirmation of thrombocytopaenia, including investigation of the peripheral blood smear.9 After also taking into account the severity of bleeding, the need for therapeutic intervention or further diagnostic testing should be assessed.9 The following case illustrates the diagnostic process for a patient who presented with haemolytic anaemia, haematuria and thrombocytopaenia.
Case study three – 17-year-old male with haemolytic anaemia, haematuria and thrombocytopaenia
A 17-year-old male presented with chronic haemolytic anaemia, proteinuria, haematuria and hypertension. Initial laboratory results showed anaemia (Hb 102 g/l, normal 130–160 g/l), raised reticulocytes (2.6%, normal 0.5–2%), thrombocytopaenia (110 x 109/l, normal 150–450 x 109/l), moderate splenomegaly (15 cm),10 enlarged kidneys with normal arteries and pronounced macroscopic haematuria.
Given the initial findings, bone marrow and kidney biopsies were performed. The bone marrow smear showed normal cell counts, with normal megakaryopoiesis, slight maturation retardation in the granulopoiesis and slightly increased erythropoiesis. This confirmed the thrombocytopaenia in this patient was not due to a production defect, but was due to platelet degradation in the circulation. The renal biopsy showed a chronic thrombotic microangiopathic nephropathy. Haemolytic uremic syndrome (HUS) and thrombocytopaenic purpura (TTP) can have a range of causes, but in this patient hyperhomocysteinaemia of three- to four-times the upper limit of normal and mild methylmalonic aciduria was suggestive of a Cobalamin C/D (Cbl-C/D) defect. Fibroblast studies and genetic testing of the methylmalonic aciduria and homocystinuria type C protein (MMACHC) gene confirmed Cbl-C defect. The patient’s nine-year-old sister was diagnosed with the same disorder at a pre-symptomatic stage.
Following diagnosis parenteral high-dose hydroxycobalamin injections (0.5 mg/kg/day) reduced the homocysteine levels, and methylmalonic acid disappeared from the urine in both the index case and his sister. Treatment addressed the haemolysis, haematuria and proteinuria and creatinine clearance has been stable for over 18 years.
The initial presentation of thrombocytopaenia with anaemia, raised reticulocytes, splenomegaly, enlarged kidneys and haematuria, exemplifies the non-specific symptomology often encountered in patients with haematological disorders. In this case bone-marrow and kidney biopsies were performed to provide further clinical clues and these identified thrombotic microangiopathic nephropathy. The diagnosis of a Cbl-C/D defect can be made by widely available blood and urine testing for homocysteine and methylmalonic acid respectively. Therefore, if testing for the more common reasons for chronic thrombotic microangiopathy have failed to identify causation, testing for Cbl-C/D defect, which is an unusual but treatable inborn cobalamin metabolism disorder, may be appropriate.11
Gaucher disease GD is an autosomal recessive inherited disease caused by mutations in the gene for glucocerebrosidase (GBA1), which is estimated to affect around 1 in 57,000 births.12 The prevalence is higher in individuals with Ashkenazi heritage (~1 in 800).1 The defect in glucocerebrosidase leads to the accumulation of glucocerebroside in the lysosomes of macrophages (Gaucher cells).3 Gaucher cells can be identified by their eccentrically placed nuclei and cytoplasm with characteristic crinkles and striations, although Gaucher cells with atypical cytomorphology have been described.13–15
GD is classified into three broad phenotypes, although clinical presentation can be highly variable, even in patients with the same type.16 Type I (nonneuronopathic) GD typically presents with splenomegaly, thrombocytopaenia, anaemia, hepatomegaly, pulmonary disease and skeletal disease (osteopenia, osteoporosis, bone remodelling abnormalities [the classic Erlenmeyer flask deformity], pathological fractures, osteolytic lesions and osteonecrosis). Rare neuronopathic variants (type 2 acute neuronopathic and type 3 chronic neuronopathic disease) demonstrate neurodegeneration in addition to the other symptoms.16
Individuals with GD have an increased risk of developing certain cancers, especially multiple myeloma and other plasma cell neoplasms, but also other haematological and lymphoid malignancies, hepatocellular carcinoma and renal cell carcinoma.17,18 GD is associated with an increased risk of cancer with a rate ratio (RR) of 2.5 (95% CI 1.1–4.7) and an increased RR of haematologic cancer of 12.7 (95% CI 2.6–37.0) compared with the general population.19 The risk of multiple myeloma is particularly high in GD with rate ratios of 51.1 (95% CI 6.2–184) compared with the general population.19 Many manifestations of GD are common to multiple myeloma, such as cytopaenia, the presence of lipid laden macrophages (pseudo Gaucher cells) in the bone marrow, monoclonal gammopathies, bone remodelling abnormalities and bone destruction due to aberrant cytokine release.1,3,20 Monoclonal gammopathy of undetermined significance (MGUS) has been reported in 19% of Gaucher patients.21 Low-risk MGUS is characterised by a monoclonal protein <15 g/l, IgG type and a normal free light chain ratio (risk of progression 1% per year).22
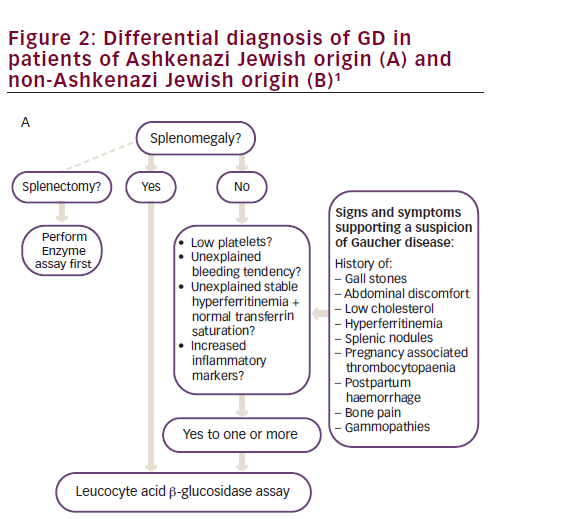
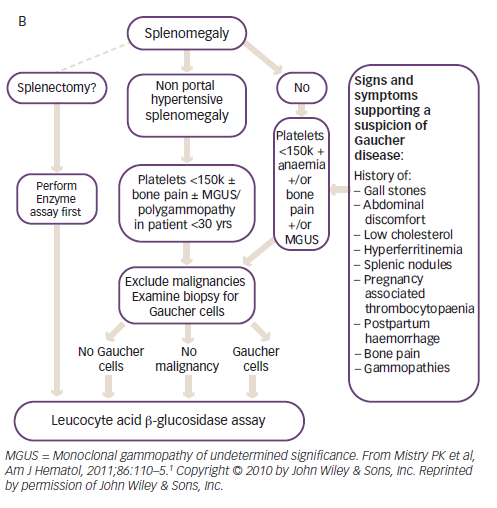
Diagnosis is delayed in approximately 25% of patients with GD due to the non-specific and heterogeneous symptoms and the rarity of the disease.1 Given that the enzyme test provides a clear diagnosis, treatment algorithms have been published to assist physicians in identifying GD (Figure 2).1 The higher prevalence of GD in individuals with Ashkenazi heritage, means that enzyme testing should be performed as a first-line investigation in anyone with such heritage presenting with splenomegaly and cytopaenia.1 The N370S mutation, a common genotype in Askenazi populations, is often associated with mild cytopaenia and splenomegaly that can escape initial detection; therefore, symptoms associated with GD such as hyperferritinaemia, low low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol, premature gallstones or osteoporosis and gammopathy should prompt the clinician to consider enzyme testing.1
Treatment
Symptomatic GD should be treated with ERT such as imiglucerase (Cerezyme®; Sanofi Genzyme, Cambridge, MA, US), velaglucerase alfa (VPRIV®; Shire HGT, Cambridge, MA, US) or taliglucerase alfa (Elelyso®; Protalix Biotherapeutics, Carmiel, Israel and Pfizer, NY, USA).23 ERT ameliorates or reverses many systemic manifestations of GD and is generally accepted as the first-line therapy for GD.5,23 However, it does not fully address all the coagulopathies in GD and therapeutic interventions should be considered to treat and/or prevent bleeding.24 In addition, some patients experience ongoing bone disease during ERT and in these patients adjunctive therapy with bisphosphonates may help improve bone mineral density.25
An alternative therapeutic approach is to use an SRT, such as miglustat (Zavesca®; Actelion Pharmaceuticals Ltd., Allschwil, Switzerland) or eliglustat (Cerdelga®; Sanofi Genzyme, Cambridge, MA, United States),26 which addresses the deficiency of the enzyme glucocerebrosidase in GD by slowing down the production of glucocerebroside. In a phase III trial, eliglustat treatment for nine months significantly improved spleen volume, haemoglobin level, liver volume and platelet count versus placebo in previously untreated individuals with GD.27 In a second, openlabel, phase III trial, eliglustat maintained haematological and organ volume stability and was non-inferior to imiglucerase in individuals with GD previously stabilised with ERT.28 Eliglustat is approved as firstline treatment for GD type 1 in Europe and the United States, giving the option of a daily oral treatment rather thanintravenous ERT infusions.29 Miglustat is indicated for the oral treatment of adult patients with mild to moderate type 1 Gaucher disease and may be used only in the treatment of patients for whom enzyme replacement therapy is unsuitable.35
The impact of Gaucher-specific therapy on the development of multiple myeloma is unknown. Polyclonal gammopathy may decrease with ERT,4 but the impact of ERT on monoclonal gammopathies is inconclusive.21,30 Follow-up of individuals with GD and monoclonal gammopathies should be performed at least every year using serum protein electrophoresis free light chains estimation and serum and urine immunofixation. If monoclonal IgG exceeds 15g/l, a bone marrow aspirate should be carried out to check for malignant transformation.31 Bone marrow aspirate and biopsy are helpful in patients with concomitant GD and myeloma in estimating the reserve of haematopoietic tissue. Induction and consolidation therapy using, proteasome inhibitors, cyclophosphamide, immunomodulatory drugs, such as thalidomide or lenalidomide, are feasible.31 Because chemotherapy may aggravate cytopaenia, ERT should be initiated in Gaucher patients with myeloma to enable treatment. Supportive therapy using biphosphonates may be appropriate.
Case study four – multiple myeloma and Gaucher disease
A 67-year-old male patient presented with bone pains, fatigue and nasal bleeding. Physical examination revealed an enlarged spleen. Laboratory investigation showed anaemia (Hb: 90 g/l, normal 130–170 g/l) and thrombocytopaenia (platelets 90 x 109/l, normal 150–400 x 109/l). WBC and differential counts were normal. Biochemical investigations showed normal renal clearance and liver function; hypercalcaemia (serum calcium 11.2 mg/dl, normal range: 8.7–10.5 mg/dl) and increased total serum proteins: 13.8 g/dl (normal 6.0–8.3 g/dl) with albumin 3.4 g/dl (normal 3.5–5.0 g/dl). Serum protein electrophoresis and immunofixation revealed a monoclonal IgGκ of 12.8 g/l. Quantitative immunoglobulin assays revealed IgG of 34.8 g/l (normal 7–16 g/l), IgM of 1.14 g/l (normal 0.4–2.3 g/l) and IgA of 0.59 g/l (normal 0.85–4.5 g/l), free light chains (FLC) κ/λ ratio 10.9 (normal 1.35–2.67). Bence-Jones protein was demonstrated in urine. Results of 24-hour urinalysis showed proteinuria (0.79 g: normal <0.15 g) with κ/λ ratio of 37.78 (normal 0.75–4.0) kappa light chain quantification 150 mg/l (normal <7.1). Serum β2-microglobulin was 8.75 (normal <2.5 mg/l). Radiography showed multiple lytic lesions in the skull, diffuse osteopenia of the femur, and the Erlenmeyer flask deformity of the distal femur. Dual energy X-ray revealed osteoporosis.
Findings suggested a diagnosis of multiple myeloma IgGκ, classified as stage IIA according to the Durie and Salmon system and stage III according to the International Staging (ISS) system.32 Bone marrow aspiration was unsuccessful (dry tap) and a trephine bone marrow biopsy was performed to confirm the diagnosis.
The splenomegaly remained unexplained. A computed tomography scan of the upper abdomen confirmed the splenomegaly (diameter 18.5 cm); there was no hepatomegaly or enlarged lymph nodes. Splenomegaly was not associated with lymphoma (no enlarged lymph nodes), chronic lymphocytic leukaemia (no lymphocytosis), or Waldenström macroglobulinaemia (no monoclonal IgM paraprotein). Differential diagnosis was considered for splenomegaly (bacterial, viral fungal or parasitic infection, liver cirrhosis, autoimmune haemolytic anaemia, spherocytosis, metabolic disease, amyloidosis etc.). The observation of the Erlenmeyer flask deformity and increased plasma chitotriosidase (16,817 nmol/ml/hr; normal <200 nmol/ ml/hr) finally led to suspicion of GD.
The bone marrow biopsy performed at diagnosis revealed Gaucher cells, which greatly replaced granulo-, erythro- and thrombopoiesis, and about 15% monoclonal plasma cells positively stained with IgG and kappa antibody. GD was confirmed by enzyme assay (low ß-glucocerebrosidase 2.6 nmol/mg prot/hr, normal 6–23 nmol/mg prot/ hr) and by genotyping (N370S/L444P).
Case study five – Gaucher disease and smouldering multiple myeloma
A 66-year-old male was diagnosed with GD when he was 45 years of age (genotype: N370S/N370S) following the identification of mild thrombocytopaenia during a routine blood count. He had a history of nephrectomy for renal carcinoma. In 2004 (aged 58), he presented with bone pain, hepatosplenomegaly, osteoporosis and a platelet count of 40–60 × 109/l (normal 150–400 × 109/l) . Laboratory investigations showed two monoclonal spikes: IgGκ 2.8 g/l and IgAκ 1.12 g/l and light chain proteinuria (Bence-Jones κ protein: 0.45 g/24h). Hb, creatinine and calcium levels were within normal values. Bone imaging showed osteolytic lesions and a well-defined mass, most probably a bone infarct or a ‘Gaucheroma’ (a mass of Gaucher cells) in the right humerus. Bone marrow aspirate and biopsy revealed 15% CD138 positive and monoclonal plasma cells with a normal karyotype on a cytogenetic study. The patient was diagnosed with concomitant GD and smouldering multiple myeloma.
Treatment with ERT and bisphosphonates was initiated. Two years later, spleen and liver volume had normalised, platelets were 100 x 109/l (normal 150-400 × 109/l), kidney function tests were normal and he was free of bone pain. After nine years, osteoporosis has improved and monoclonal spikes and light chain proteinuria are stable.23
The risk of multiple myeloma is particularly high in GD and both diseases share common symptoms.1,3,19,20 These two cases highlight the importance of considering early detection of MGUS, multiple myeloma or amyloidosis in individuals diagnosed with GD. Additionally, GD should be considered in relatively young patients diagnosed with multiple myeloma, smouldering myeloma or MGUS. The diagnosis and treatment of concomitant GD and myeloma is challenging. Immunophenotyping and cytogenetic testing of bone marrow specimens are essential for the diagnosis of myeloma in a marrow infiltrated with Gaucher cells. ERT should be initiated in GD patients with myeloma in order to prevent aggravation of cytopaenia due to chemotherapy. A search for monoclonal gammopathy should be systematically (and periodically) done in patients with GD given the increased susceptibility to plasma-cell neoplasm.
Case study six – warfarin-like poisoning
A 56-year-old woman was investigated for bruising and recent gum and skin bleeds. The patient had no relevant medical history, was not taking any medications and had a normal diet. Following clinical investigation, an isolated prolongation of prothrombin time was detected, and deficiency of all vitamin K-dependent clotting proteins. Only after a third interview, when the patient indicated that she used rat poison to prevent the rats from eating the food for the hamsters but forgot to wash her hands before having lunch, did the doctor realise that the woman’s bruising could be related to warfarin-like poisoning and the effects of vitamin K antagonists.
An overview of vitamin K antagonists
Vitamin K antagonists were first identified in 1941 and while the potential clinical value was recognised from the outset, the synthesis of the vitamin K antagonist warfarin was first exploited as a rodent poison. Vitamin K antagonists interfere with the synthesis of vitamin K-dependent proteins, including the clotting proteins prothrombin factor II, VII and X, and have major anti-thrombotic effects.33 In this case the exposure was accidental as the patient was using warfarin as a rodenticide to deal with her rat infestation.
Key clinical signs
The most sensitive test for the effect of vitamin K antagonists is the prothrombin time, sensitive to the levels of three of the four vitamin K-dependent clotting proteins (VII, X and prothrombin). The International Normalised Ratio (INR) is a form of standardised prothrombin time. The prolongation of the partial thromboplastin time is usually not as severe as the prothrombin time.
Warfarin at a dose of 5–10 mg a day induces severe depletion of factor VII in one to two days. Shortly thereafter, factor IX, X and prothrombin levels will also fall. Lower doses will also result in the same low levels but after a longer period of time. The regular inadvertent intake of vitamin K antagonists (as in the exposure to rat poison) is seldom reported. The clinical effect depends on the vitamin K intake of the individual.
Treatment ,br>
The effects of vitamin K antagonists are counteracted by administration of extra vitamin K.34 In general, 10 mg per day for a few days will be sufficient (depending on the t1⁄2 of the causative compound). In the case of acute severe bleeding, clotting proteins may be administered. ,/p.
Summary
The recognition of rare haematological diseases is challenging. Given that these diseases often initially present with fairly non-specific haematological symptomology, haematologists are likely to face these challenging diagnoses in their clinical practice. Therefore, it is important for haematologists to have an awareness of rare haematological diseases and to consider them when making their initial differential diagnoses based on information obtained from the patient, the clinical findings and laboratory values. As the cases reviewed here show, once the possibility of a rare haematological disease has been identified, confirmatory testing is relatively straightforward in the majority of cases. In addition, as effective therapeutic interventions are available for many of these conditions, it is important that patients are not denied effective treatment due to diagnostic delays. Over the last 10 years, the Sherlock Holmes symposia have raised the awareness of rare haematological diseases among thousands of haematologists around the world. Through the greater disease awareness promoted by these symposia, it is hoped that many patients already have and will benefit from earlier recognition and treatment for their rare haematological diseases.












