An ever-expanding understanding of the biological basis for multiple myeloma (MM) combined with a widening array of effective therapies has dramatically improved outcomes for patients and has provided new opportunities for disease segmentation and targeted therapeutics.1 While this progress is most welcome, it has created unique challenges for clinicians because of the spatial and temporal heterogeneity, of the disease.2 These features make it difficult to determine prognosis for any given patient and render the selection of a rational sequence of therapeutic interventions bewildering. Here we provide a panoramic view of recent progress in understanding the genetics of MM and its potential to influence therapeutic decision-making.
Multiple myeloma molecular segments
MM results from an abnormal clonal proliferation of plasma cells.3 In non-hyperdiploid MM, the majority of molecular events are a result of abnormal class switch recombination, which results in translocations involving the 14q32 region (the immunoglobulin heavy chain locus).4 The remainder of newly diagnosed MMs are hyperdiploid cases associated with trisomies of the odd-numbered chromosomes but, in practice, there is some overlap between these two disease categories.5,6 Analysis of the genetic makeup of MM shows both heterogeneity between cases but also within the same case at different sites. It is this sub-clonal heterogeneity that leads to variable prognosis and differential responses at distinct anatomic sites. Interestingly, copy number and structural events are common in MM and are prognostically critical.3,7 Mutational changes in within the coding sequences are also prevalent, with a limited number of recurrent mutations that retain a long tail of low-frequency events. The most common mutations impact KRAS, NRAS, and BRAF, with an excess of inactivating mutations in TP53 and FAM46C. Up to 50% of patients have activation of the RAS/MAPK pathway, whereas 12% have inactivating mutations of the NF-κB pathway.8 Oncogene mutations are frequently more clonally dominant than mutations in tumor suppressor genes. Acquired genetic variants lead to changes in protein expression and are responsible for the inter-patient differences in clinical outcome. They can thus be used to both segment the disease and to define prognosis.
Risk stratification is clinically important in disease segmentation and, in recent years, the International Myeloma Working Group has integrated chromosomal abnormalities into traditional prognostic models.9,10 In addition to the conventional laboratory criteria of the international staging system (ISS), the revised ISS score (R-ISS) incorporates the presence of t(4:14), t(14;16), and del(17p13) to identify patients with inferior survival. The constellation of ISS III disease, combined with cytogenetic abnormalities by fluorescence in-situ hybridization, predicted an overall survival of 33% at 4 years, compared to 76% in a group defined by ISS I disease and neither prognostic marker.11 The R-ISS, however, lacks some specificity for adverse outcomes, especially among intermediate-risk patients in certain populations.12 The UK Medical Research Council group and the Institute for Functional Medicine have incorporated genetic markers into this system and have produced models incorporating the t(14:16) as well as chromosome 1q gain to improve the risk stratification.13,14 It is now accepted that cytogenetic groups at higher risk include any of the t(4:14) translocations, amplification of 1q, and loss of the short arm of chromosome 17. Ultra-high-risk groups contain more than one of these abnormalities.
A more specific way of defining an ultra-high-risk group has recently been described based on mutation patterns. These patients have been identified as harboring “double hit” MM, a label that is based on bi-allelic inactivation of P53 or ISS III and amplification of 1q.8 This group makes up some 6% of newly diagnosed MM and has a very poor outcome. Chimeric antigen receptor T (CAR T) cells are being evaluated in clinical trials for these cases.15 Further risk groups can be identified by incorporating gene expression and mutation analysis. This approach is able to identify a group of patients with more proliferative disease and correspondingly impaired outcomes. Attempts to identify more mutations that were both prevalent and exerted a significant risk were unsuccessful, suggesting a need to find new mechanisms and mutations within the non-coding sequences.
Targeted therapies for multiple myeloma
There has been a very gratifying expansion in targeted agents with specific mechanisms of action that can be split into a number of functional categories. This progress has facilitated personalized therapy, where a diagnostic approach is used to identify cases where a specific agent can be best used.
Immunomodulatory drugs
Immunomodulatory agents were the earliest class of targeted therapeutic agents used in MM and were commonly used prior to the delineation of their mechanism of action. More recent investigations have shown that they exert their activity via modulation of the activity of cereblon, impacting the targeting of ikaros and aiolos to the proteasome for degradation.16,17 Through this mechanism, blockade of cereblon is directly toxic to plasma cells via down-regulation of IRF4 and MYC and also serves to enhance the immune response by increased production of interleukin (IL)-2 in natural killer (NK) cells.18 A number of new drugs are in development that promise to further augment the activity of immunomodulatory agents.
Proteasome inhibitors
Proteasome inhibitors emerged as a viable therapy in relapsed refractory MM (RRMM) and rapidly moved to first-line therapy through a series of phase II and phase III trials.19,20 These agents selectively target the proteasome, leading to an accumulation of ubiquitinated proteins within the cell. As plasma cells are highly synthetically active and require active modulation of the misfolded protein response to avoid endoplasmic reticulum stress, disruption of this process is particularly cytotoxic in plasma cell neoplasms. Disruption of NF-κB signaling, a pathway that is activated by mutations commonly seen in MM, has also been implicated as a potential mechanism.2
Anti-CD38 antibodies
Daratumumab is a first-in-class anti-CD38 antibody that is highly active in combination with immunomodulatory drugs and proteasome inhibitors. The agent has a number of mechanisms of action, including Fc receptor-mediated crosslinking of CD38 with subsequent downstream induction of apoptosis.21,22 It also mediates antibody-dependent cellular cytotoxicity and depletes regulatory T cells, enhancing the immune response against plasma cells. In the SIRIUS trial, a phase II study in RRMM, single-agent daratumumab had an overall response rate (ORR) of 29% and a progression-free survival (PFS) of 3.7 months.23 It was then evaluated in combination with a proteasome inhibitor (CASTOR) and an immunomodulator (POLLUX), with excellent results, including a significant number of minimal residual disease negative complete responses.24,25 In the first-line setting, the ALCYONE and MAIA studies have led to its approval in patients not eligible for transplant.26,27 The GRIFFIN study, which is presently ongoing, is evaluating daratumumab in combination with bortezomib, lenalidomide, melphalan, and dexamethasone for the upfront treatment of transplant-eligible patients.26
Anti-SLAMF7 antibodies
The first approved agent in this class of therapies is elotuzumab, an immunostimulatory IgG1 monoclonal antibody that targets signaling lymphocytic activation molecule family member 7 (SLAMF7), a surface glycoprotein expressed on NK cells, as well as aberrant plasma cell clones.28–30 Although elotuzumab has no single-agent activity, it has demonstrated improved outcomes in a phase III setting when combined with lenalidomide and dexamethasone (ELOQUENT-2 trial), with a superior ORR (79% versus 66%; p<0.001) and favorable PFS (19.4 versus 14.9 months; hazard ratio [HR] 0.70; p=0.001) as compared to standard doublet therapy.31
Anti-apoptotic therapy
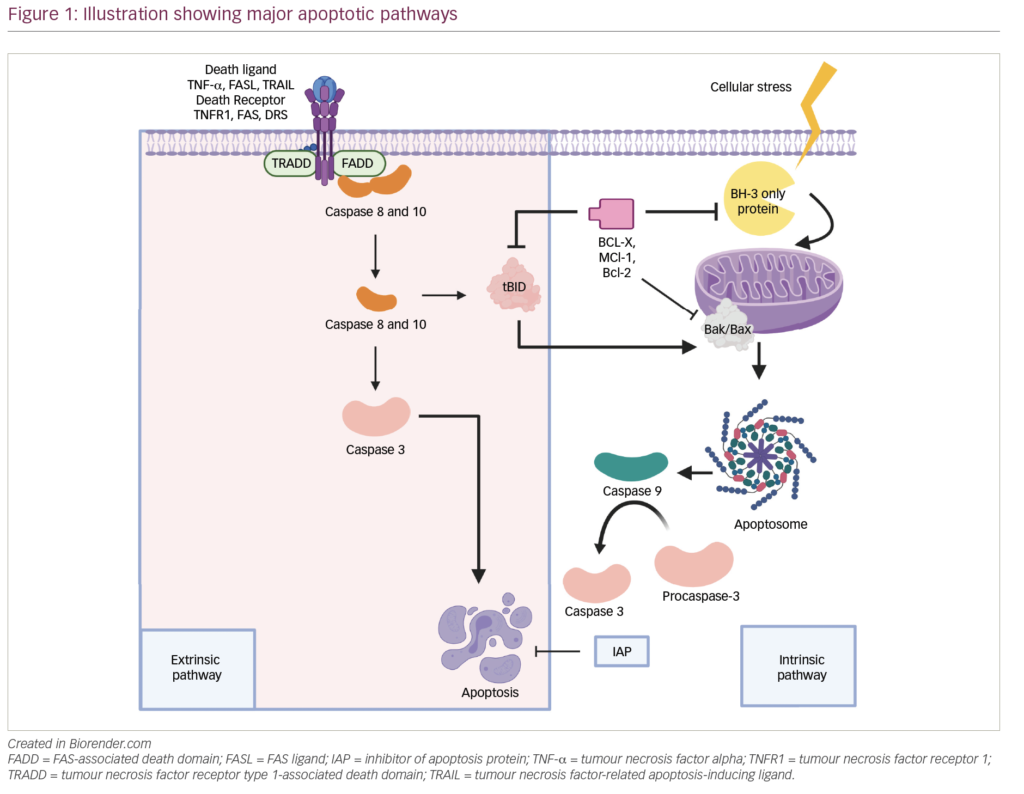
Venetoclax, a small molecule BH3 mimetic, inhibits BCL-2 as a means of inducing apoptosis and has proven to be effective in a variety of B-cell neoplasms.32 As a single agent, venetoclax produced the highest response rates in t(11;14) cases.33 The drug has also demonstrated increased efficacy in combination with bortezomib, possibly through knockdown of mcl1.34 The phase III BELLINI trial evaluated venetoclax, bortezomib, and dexamethasone versus bortezomib and dexamethasone alone. This trial enrolled 291 patients and, at a median follow up of 18.7 months, and demonstrated an enhanced PFS (22.4 versus 11.5 months; HR 0.630; p=0.01) favoring the venetoclax arm. The ORR (82% versus 68%; p<0.01) and the proportion of patients achieving a very good partial response (VGPR) or better (59% versus 36%; p<0.01) were also very encouraging.35 However, the study was placed on a clinical hold by the US Food and Drug Administration (FDA) after an increase in the rate of death (21.1% versus 11.3%; p<0.01) was noted in the venetoclax arm.36 The cause for these excess deaths is the subject of intense investigation as this drug constitutes a very important step forward, especially for cases harboring the t(11;14) translocation.
Histone deacetylase inhibitors
Histone deacetylase inhibitors (HDACs) were identified as potential targets across multiple tissue types due to their widespread overexpression in neoplastic tissue and have been successfully targeted in several lymphoid neoplasms.37 In addition to the modulation of chromatin structure, HDACs may play an active role in the handling of misfolded proteins and thus represent attractive targets for anti-myeloma therapy.38 Early evaluation of vorinostat, a pan-HDAC inhibitor, in RRMM showed limited efficacy and, in many cases, an unacceptably narrow therapeutic index.39 Panobinostat, a second-generation HDAC inhibitor, was evaluated in combination with bortezomib and dexamethasone in RRMM in the PANORAMA 1 and 2 and subsequent phase III PANORAMA 3 trials. It showed a significant benefit that was most obvious in a pre-specified subset of high-risk disease and was approved for clinical use.40 These benefits were mitigated to a certain extent by high rates of severe gastrointestinal side effects, which have led to further regulatory guidance regarding this class of agents.41
B-cell maturation antigen targeted therapies
The B-cell maturation antigen (BCMA) is ubiquitously expressed on plasma cells and is a good target for T-cell fixing therapies, including CAR-T and bispecific antibodies.42,43 CAR T cells targeting this antigen have excellent response rates in RRMM. Two of the most advanced products have demonstrated response rates up to 80% with associated PFS of 11 months, which is remarkable in this setting and raises expectations for a very significant impact in newly diagnosed MM.44,45 Early-stage evaluation of the antibody–drug conjugate, GSK911, and bispecific agents show similar response rates, implying that we will have significant clinical work to evaluate the most appropriate setting for such therapies.
Selinexor
Selinexor is an oral inhibitor of exportin 1, the gene product of XPO1, which is a transport protein involved in the nuclear export of tumor suppressor proteins and the glucocorticoid receptor.46,47 Inhibition of this protein may increase intra-nuclear activity of these tumor suppressors and prevent aberrant myeloma cells from escaping apoptotic signals as well as cell cycle checkpoints.47 In refractory cases, a significant clinical benefit was seen leading to its approval.48 The clinical uptake of the drug will depend upon the tolerability of its gastrointestinal side effects.
Matching therapies to molecular segments
With a range of different molecules with distinct modes of action, the clinical challenge is how to optimize their application, both in terms of their sequence of use and the molecular segment to which they are applied. The targeted agents were developed to have broad activity but, as a community, we will require the introduction of disease segmentation to maintain therapeutic progress. Only by doing this will we be able to design efficient clinical trials that are of a size and cost that is practical. This is not really a giant step as personalization of therapy in the clinical setting is a reality, with frailty scores being widely used to determine therapeutic strategies in older patients.49 Recent clinical trial data have led to a change in approach for transplant non-eligible patients, based initially on the results of the FIRST study which showed the benefit of lenalidomide, and subsequently the MAIA trial, which further optimized its use in the combination with daratumumab, which is now a standard of care in this group of patients. The outstanding question is whether proteasome inhibition can be integrated into the treatment approach for elderly patients with any potential increased toxicity being offset by deeper responses and potentially improved survival.27
In younger patients, risk stratification is an area where there is a clear role for disease segmentation and the development of specific therapeutic strategies. As defined above, we have made significant progress in defining molecular testing strategies able to identify high-risk cases in the clinic. A review of the impact of therapy over time suggests that, in contrast to low-risk disease, we have not made significant improvements in outcomes for very aggressive disease.50 However, some lack of clarity remains over the use of the term “high-risk”, and here we specifically refer to very aggressive disease that is proliferative and tends to relapse early after treatment. Future development of therapies for high-risk MM should rely on systematically performing sequential trials to improve outcomes. The utilization of immune active agents such as daratumumab and T-cell targeting constructs have clear potential to improve the outcome of this group; however, we should not expect cures for this segment in early applications of these agents. Instead, we should anticipate steady improvements in outcomes over time as the most appropriate sequence of therapy is defined. In contrast to high-risk disease, we have made significant impacts on the outcome of low-risk MM with significant cure fractions emerging at 10 years and beyond. The challenge in this segment is to be able to reliably identify patients at presentation and to move from strategies aimed at disease control to strategies based on maximizing long-term survival and cure.
Analyses of sets of patients with molecular risk factors have led to the conclusion that proteasome inhibitors have a particular impact in the t(4;14) molecular segment.51 Patients harboring high-risk genetic alterations, including t(4;14), t(14;16) and del(17p) that were treated with melphalan, prednisolone, and bortezomib had clinically meaningful improvements in time to progression, complete response rates, and overall survival.52 A number of additional studies have consistently demonstrated this benefit for t(4;14); however, laboratory and clinical analysis of the t(14;16) segments failed to show a substantial benefit for the use of proteasome inhibition.53 Both lenalidomide and daratumumab have been suggested to lack activity in high-risk disease. However, closer review of data from recent trials of both agents showed clear activity in these settings and, while there may be an inferior hazard ratio in the more aggressive disease, this is what would be expected in higher risk presentations.26
Other primary molecular segments may benefit from the use of specific agents. While the Bellini study showed potential benefits of bortezomib and venetoclax across all subgroups, the single agent venetoclax analyses were overwhelmingly positive in the t(11;14) segment.33 Thus, from a clinical perspective, trials exploring venetoclax in the t(11;14) subgroup offer an exciting path forward and will provide a focus for effective treatment of this disease segment. On a more speculative note, the immunomodulatory imide drugs may have a specific benefit in the hyperdiploid segment, in which deregulation of MYC is common.54
Activation of the MAPK pathway by mutation of RAS and BRAF and by other mechanisms presents a clear pathway for testing the role of mutational targeted therapy in MM. In this respect, the use of MEK and BRAF inhibitors represents an imperfect, but nonetheless valid, approach for testing whether this strategy offers any clinical utility. The application of these agents showed clear activity in cases with mutations but were complicated by early relapse as a consequence of the sub-clonal nature of the target mutations.55,56 Despite these findings, strategies for the inhibition of this pathway represent a clear pathway forward for therapy in MM. Likewise, the NF-κB pathway remains a clear pathway that can be specifically manipulated.
The introduction of mutation-targeted approaches into the clinic will require significant changes to the infrastructure required for both clinical trials and the delivery of routine clinical care. Molecular diagnostic panels will form an integral part of clinical management and must be regularly available to identify risk status. Furthermore, the identification of immune segments where the value of immune active agents targeting BCMA can be most appropriately utilized will require the widespread use of flow cytometry. All of these changes will require strong academic centers working with diagnostic pathologists and regional colleagues to devise appropriate care pathways that optimize patient access and convenience.