Multiple myeloma (MM) is the second most common haematological malignancy, affecting an estimated 450,600 patients worldwide, with an estimated 34,920 new diagnoses and 12,410 patient deaths in the USA in 2021.1,2 Although established curative therapies for MM have not yet been defined, we have observed an incremental increase in survival, with a doubling of 5-year survival rates owing to the advent of novel therapeutics.3,4 Based on extrapolations from available population-based databases, it seems reasonable to surmise that a patient with newly diagnosed MM, without underlying comorbidities, will have an average overall survival (OS) of more than 10 years.4 The approval of bortezomib, in 2003, represented the first novel therapy to be approved, and since then an additional dozen drugs with various novel mechanisms of action, including immunotherapies, have been approved.5 Most recently, the US Food and Drug Administration (FDA) approved the first cellular therapy for myeloma, the chimeric antigen receptor (CAR) T-cell therapy, idecabtagene vicleucel.6
Over the last 10 years, the advancement of novel treatments for MM has been astonishing; two decades ago, life expectancy was dismal, given the level of toxicity and relative ineffectiveness of traditional cytotoxic chemotherapy.7 The first intervention that changed the tide was the development of high-dose melphalan chemotherapy and autologous stem cell support (HDM-ASCT).8 Initial trials of HDM were associated with improved efficacy over standard-dose cytotoxic regimens, but at the expense of bone marrow toxicity, leading to delayed recovery time and associated complications.9,10 It was ASCT that, in combination with HDM, led to decreased toxicity and improved outcomes, supporting the further development of this approach by Barlogie, McElwain and others in the mid-1980s.11,12
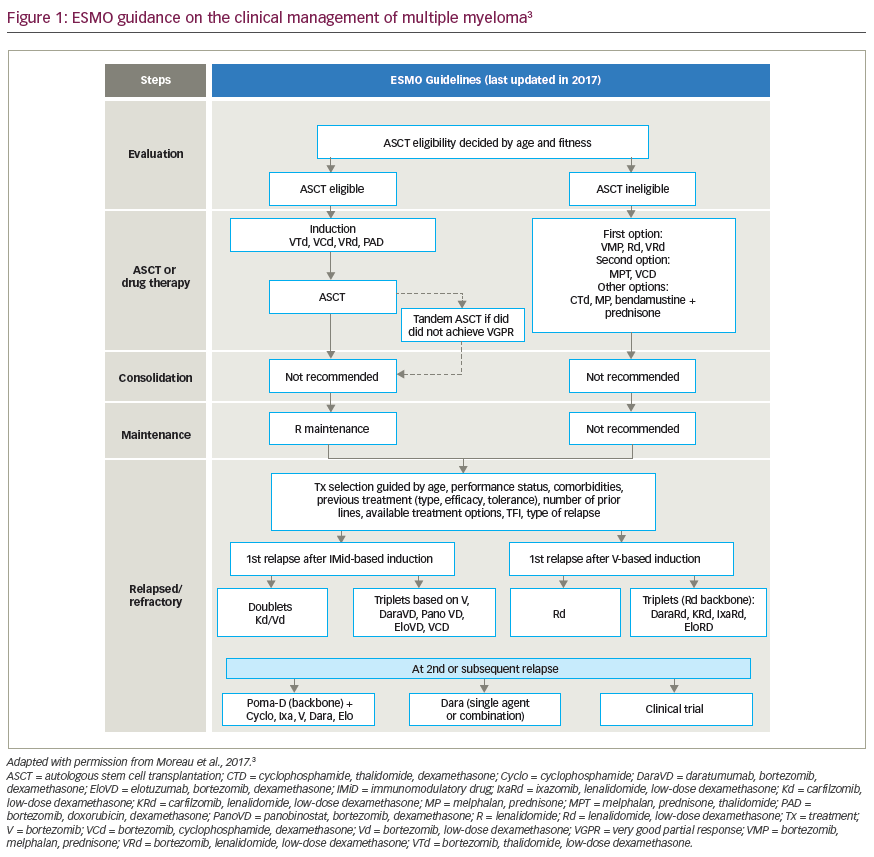
At present, the standard first-line treatment for newly diagnosed MM includes novel proteosome inhibitors and immunomodulatory-drug triplet-based regimens, which have shown effective and sustained minimal residual disease (MRD)-negative remissions.13,14 Most recently, clinical trials have added the anti-CD38 monoclonal antibody, daratumumab, on top of novel triplet backbone combinations, yielding high rates of MRD negativity (up to 71%) and high rates of progression-free survival (PFS), in the absence of HDM-ASCT.15,16 These exponential improvements in efficacy with novel triplet- and quadruplet-based therapies have changed the treatment paradigm for MM, and raised questions regarding the value of first-line HDM-ASCT.17 However, in patients who achieve early MRD-negative remission after stem cell harvest, delaying HDM-ASCT until the time of relapse is a favourable approach to reduce both early and delayed toxicity. Therefore, melphalan chemotherapy is likely to remain in the MM physician’s treatment toolbox, for now, for various specific indications. As patients with MM live longer and receive subsequent lines of therapy, there remains an unmet need for available drugs in the heavily pretreated, relapsed and refractory MM (RRMM) setting.
The recent development, and ultimate FDA approval, of melphalan flufenamide (melflufen; Pepaxto®, Oncopeptides, Inc. Waltham, MA, USA) is an example of taking an established active MM chemotherapy (i.e. melphalan) and modifying it to be more targeted with regard to drug delivery. It is a peptide–drug conjugate that, by interacting with aminopeptidases, releases the active alkylating agent specifically into cancer cells, avoiding normal cells. In our opinion, melflufen is a respectable addition to the MM clinician’s armamentarium in the treatment of patients with RRMM requiring multiple subsequent lines of therapy. In this review, we summarize the biological rationale, preclinical studies, early-phase clinical studies and the pivotal trials leading to the approval of melflufen for the treatment of RRMM, and describe ongoing ancillary and supportive clinical studies.
Preclinical overview of melflufen
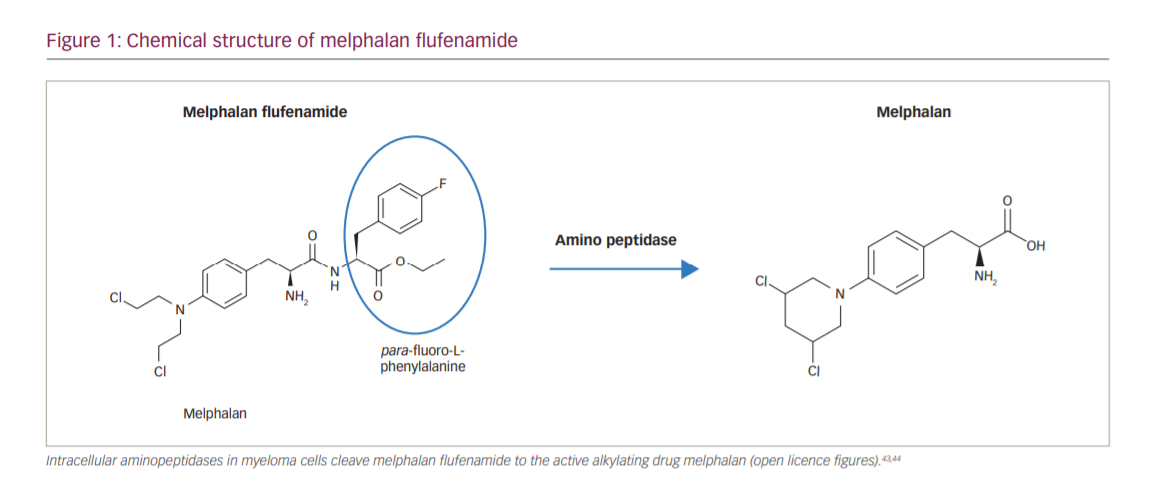
Melflufen consists of melphalan conjugated to the peptide para-fluoro-L-phenylalanine, creating a lipophilic drug (Figure 1).18 Once melflufen enters myeloma cells, cytotoxic activity is dependent on the cell’s aminopeptidase N activity to cleave the active alkylating agent. The intracellular accumulation of melflufen is thought to occur rapidly over the cell membrane by lipophilic passive diffusion. Once the prodrug has accumulated intracellularly, the formation of the active drug is dependent on high expression of aminopeptidases, which have been observed in MM and other malignant histologies.18 Therefore, the antitumour activity of melflufen is less dependent on exposure time and half-life than melphalan, and in vitro, peak activity is observed after just 30 minutes.
In both in vitro and in vivo model systems, melflufen has been shown to be more potent than melphalan, possibly deploying a more efficient therapeutic strategy for delivering higher concentrations of intracellular melphalan, and in human plasmacytoma xenograft mouse models, treatment with melflufen significantly inhibited MM tumour growth and prolonged survival compared with melphalan.19,20 With regard to toxicity, transient haematological toxicity and end-organ toxicities of the testes, epididymides, bone marrow and spleen were observed in animal studies.21
Early-phase clinical studies
The first-in-human phase I/IIa study of melflufen was in patients with advanced solid tumours and no available treatment options.22 Once the initial safety profile was characterized, melflufen was evaluated in the O-12-M1 study, a single-arm, multicentre, dose-expansion and confirmation phase I/II study in patients with RRMM who had received at least two prior lines of therapy.23 In phase I, a maximum tolerated dose of melflufen 40 mg every 21 days plus weekly dexamethasone was established; this was extended to a 28-day cycle in phase II to prolong haematological recovery time. Of the 45 patients who received this combination in phase II, the overall response rate (ORR) was 31%, with nine patients achieving a partial response (PR) or better with a median duration of response (DoR) of 8.4 months. Commonly reported adverse events (AEs) included thrombocytopenia, neutropenia, anaemia, pyrexia, asthenia, fatigue, nausea and diarrhoea.
Pivotal trials and FDA approval
The HORIZON study
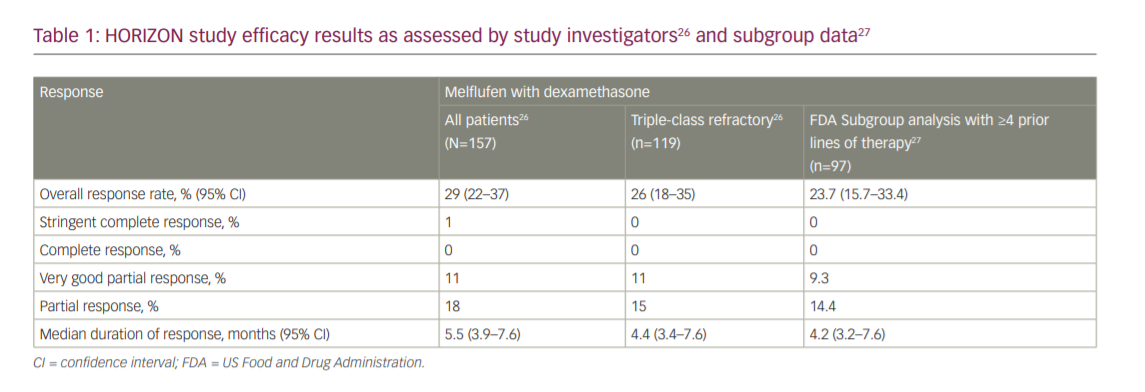
On 26 February 2021, the FDA granted accelerated approval to melflufen in combination with dexamethasone for patients with RRMM who have received at least four prior lines of therapy and whose disease is refractory to at least one proteasome inhibitor, one immunomodulatory agent and one CD38-directed monoclonal antibody, based on the results of the HORIZON study (OP-106; NCT02963493).24,25 The HORIZON study was a single-arm, multicentre, phase II study in patients with RRMM refractory to pomalidomide and/or an anti-CD38 monoclonal antibody.26 Eligible adult patients had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0–2, RRMM with disease progression and measurable disease, and had received ≥2 prior lines of therapy (including an immunomodulatory agent and proteasome inhibitor, and were refractory to pomalidomide and/or an anti-CD38 monoclonal antibody). Patients received melflufen 40 mg as a 30-minute central intravenous infusion on day 1 of each 28-day cycle in combination with dexamethasone 40 mg oral weekly until disease progression or unacceptable toxicity, or at the discretion of the physician/patient. The primary endpoint was ORR, per the International Myeloma Working Group response criteria, as assessed by the investigator and confirmed by an independent review committee (IRC). Secondary endpoints included DoR, PFS, OS and safety. The study was designed to enrol 150 patients with the statistical hypothesis that assumed an ORR of 30% with an exact 95% confidence interval (CI) of 23–38%.26
At the data cut-off date, 83% of the 157 study-treated patients had discontinued treatment mostly due to disease progression (56%) or AEs (17%). The median duration of treatment was 3.8 months. Patients had a median age of 65 years and received a median of five prior lines, 98% were refractory to their last line of therapy, 76% had triple-class refractory disease, 38% had high-risk cytogenetics, and 25% had an International Staging System stage III disease.26
The HORIZON study reported an ORR per investigator assessment of 29% (95% CI 22–37%), a median time to response of 1.9 months and a median DoR of 5.5 months (95% CI 3.9–7.6 months), with one patient achieving a stringent complete response (CR), 17 a very good PR (VGPR) and 28 a PR. In the triple-class refractory subgroup, the ORR was 26% (95% CI 18–35%) and median DoR was 4.4 months (95% CI 3.4–7.6 months), with 13 patients achieving a VGPR and 18 a PR. The ORR per the IRC was 30% overall (95% CI 23–38%) and 26% in the triple-class refractory subgroup (95% CI 18–35%). Median PFS was 4.2 months (95% CI 3.4–4.9 months) overall and 3.9 months (95% CI 3.0–4.6 months) in the triple-class refractory subgroup. Median OS was 11.6 months (95% CI 9.3–15.4 months) overall and 11.2 months (95% CI 7.7–13.2 months) in the triple-class refractory subgroup (Table 1).26
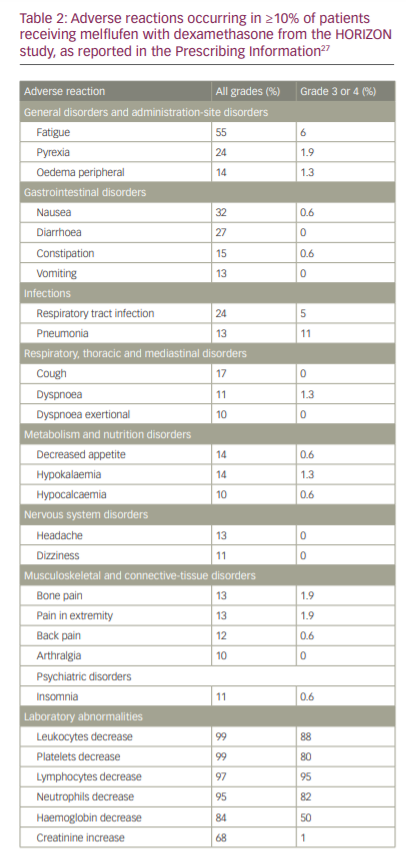
Treatment-emergent AEs of any grade were observed in all patients, with common non-haematological AEs being nausea (32%), fatigue (29%), asthenia (27%), diarrhoea (27%), pyrexia (24%), cough (17%), upper respiratory tract infection (16%), constipation (15%), decreased appetite (14%), hypokalaemia (14%), peripheral oedema (14%), headache (13%), vomiting (13%), bone pain (13%), pain in extremity (13%) and pneumonia (13%). Grade ≥3 AEs occurred in 150 patients (96%), with the most common being neutropenia (79%), thrombocytopenia (76%), anaemia (43%), pneumonia (10%) and hypophosphataemia (5%). Serious AEs occurred in 77 patients (49%), most common of which were pneumonia (9%) and febrile neutropenia (5%) (Table 2).26
The results from the pivotal HORIZON study were the basis for the submission of a new drug-licensing application for registration to the FDA, which granted melflufen priority review status and a Prescription Drug User Fee Act action goal date of 28 February 2021.25 Melflufen was granted accelerated approval based on determining a favourable benefit–risk profile. In the FDA executive review, approval was based on the ORR and DoR from a subgroup analysis of 97 patients in the HORIZON study who received ≥4 prior lines of therapies and were refractory to a proteasome inhibitor, immunomodulatory drug and an anti-CD38 monoclonal antibody. The ORR per investigator assessment in this subgroup was 23.7% (95% CI 15.7–33.4%), and the median DoR was 4.2 months (95% CI 3.2–7.6 months) (Table 1).
In terms of safety, the FDA Prescribing Information describes the following adverse reactions: thrombocytopenia (grade 3, 26%; grade 4, 54%), neutropenia (grade 3, 41%; grade 4, 40%; febrile neutropenia, 6%), anaemia (grade 3, 50%), infection (grade 3, 20%; grade 4, 1.9%; fatal infection, <1%), increased risk of mortality with dosages higher than the recommended dosage, secondary malignancies (myelodysplastic syndromes or acute leukaemia) and embryo-foetal toxicity.27
The OCEAN study
Due to the lack of a randomized control arm in the HORIZON study, FDA accelerated approval was based on a surrogate endpoint (ORR) thought to predict clinical benefit in treating a serious disease with unmet medical need (RRMM), as is standard protocol.28 However, accelerated approval requires the sponsor to confirm clinical benefit by submitting a final study report and dataset from a randomized phase III trial, which, in this case, should verify the clinical benefit of melflufen in patients with RRMM. Patients in said trial should be randomized to receive either melflufen or standard therapy. The primary endpoint should be PFS assessed by an IRC, and secondary endpoints should include ORR and OS.21
Final report submission to the FDA is scheduled for February 2022, and will comprise the ongoing, confirmatory, phase III, randomized OCEAN trial (OP-103; NCT03151811), in 495 patients who have received 2–4 prior therapies and are refractory to lenalidomide. Patients have been randomized to receive dexamethasone in combination with either melflufen or pomalidomide. The primary endpoint is PFS, and secondary endpoints are ORR, DoR and OS.25,29 However, on 8 July 2021, the FDA requested a partial clinical hold on all melflufen studies, based on updated results from the OCEAN study following an IRC reassessment, which was missing clinical data on 29 patients.30 In the new analysis, melflufen met the primary endpoint of superior PFS compared with pomalidomide (hazard ratio [HR] 0.79; 95% CI 0.64–0.98; p=0.0311) as determined by the IRC. However, the key secondary endpoint of OS had an HR of 1.10 (95% CI 0.85–1.44) in favour of pomalidomide for the intention-to-treat population. Further investigation is ongoing and will include analyses of subgroups that might have driven the detrimental OS. On 22 October 2021, however, Oncopeptides, the manufacturer of melflufen, after discussion with the FDA, withdrew the product from the US market. Therefore, it is no long available to the general MM population, but patients who were previously receiving it and deriving benefit will be able to continue treatment through a compassionate use program with Oncopeptides.
Other clinical trials evaluating melflufen
Melflufen triplet combinations
The ongoing, two-arm, phase I/II ANCHOR study (OP-104; NCT03481556) is evaluating triplet regimens of melflufen (30 or 40 mg) plus dexamethasone in combination with either bortezomib or daratumumab in patients with RRMM, who have received 1–4 prior lines of therapy, and are refractory to an immunomodulatory drug and/or a proteosome inhibitor.31,32 In preliminary results, amongst 33 patients in the daratumumab arm, ORR was 70% (1 stringent CR, 1 CR and 10 VGPR), with a median DoR of 12.5 months, and a median PFS of 11.5 months.32 In the bortezomib arm (n=13), ORR was 62%, with 1 CR and 4 VGPR.32 Furthermore, the daratumumab triplet combination is being compared with daratumumab alone in the phase III, randomized LIGHTHOUSE trial (OP-108; NCT04649060), in patients with RRMM previously treated with an immunomodulatory drug and a proteasome inhibitor. Patient enrolment has begun, with 240 patients planned to be enrolled; the primary endpoint is PFS and key secondary endpoints are ORR, DoR and OS.33,34
Clinical pharmacology trials
The ongoing, phase II BRIDGE study (OP-107; NCT03639610) is evaluating the pharmacokinetics of melphalan during treatment with dexamethasone in patients with RRMM and moderate to severely impaired renal function.35,36 Renal and hepatic insufficiency are not expected to alter pharmacokinetics significantly, given that the metabolism of melflufen is dependent on cellular aminopeptidases and melphalan is metabolized by spontaneous hydrolysis in the blood.35 The study is ongoing, with seven of the 35 planned patients thus far enrolled.27,37
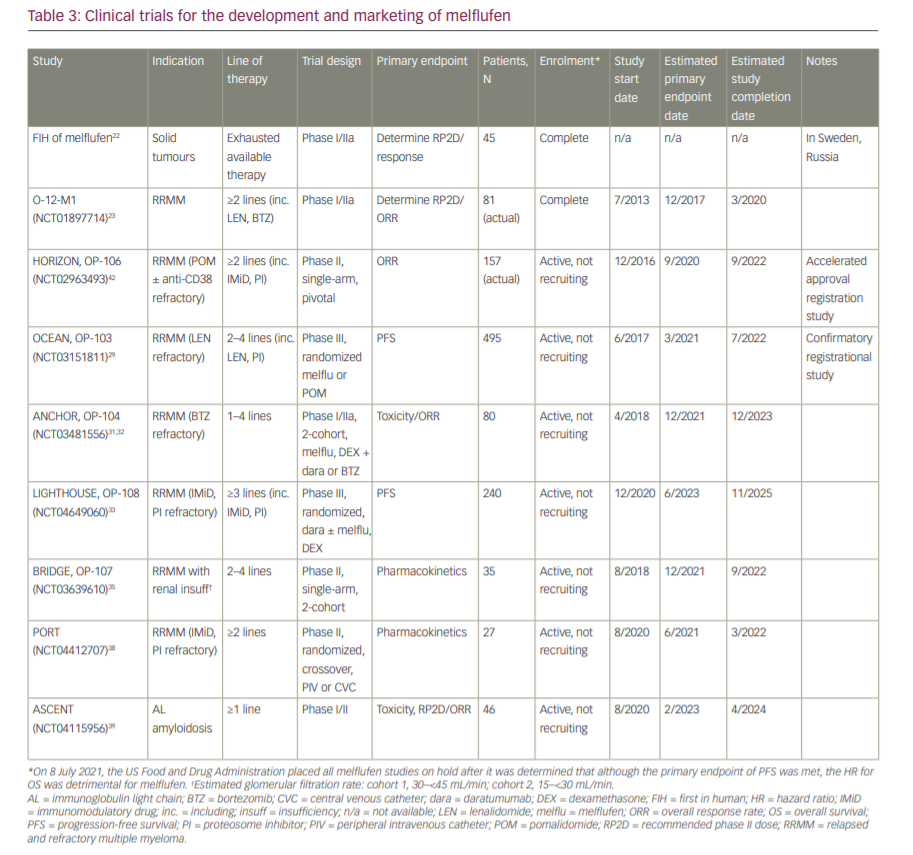
The current approval of melflufen is based on administration by central intravenous infusion, which makes it less practical than standard peripheral intravenous infusions for patients. The PORT study (NCT04412707), a randomized, two-period, cross-over, phase II study, has not yet been activated, but will be recruiting up to 27 patients to compare the pharmacokinetics, safety and tolerability of peripheral versus central intravenous administration of melflufen.38 Finally, in the phase I/II ASCENT study (NCT04115956), 46 patients with immunoglobulin light-chain amyloidosis following one line of therapy will be enrolled to evaluate the safety and efficacy of up to eight cycles of melflufen with dexamethasone.39 Table 3 summarizes the melflufen drug development pipeline.
Conclusions and future perspectives
The melflufen drug development programme is a prime example of how an older cytotoxic drug is not just recycled in the context of a changing field towards novel immunotherapies, but also improved by making it more targeted and introducing as a later line of therapy. As triplet and now quadruplet novel therapy combinations have dominated the treatment of newly diagnosed MM, achieving high rates of MRD-negative remission, HDM-ASCT is moving from an early treatment modality to one that is used in the relapsed setting. This is critical, as independent investigators are now showing the detrimental, long-term, mutagenic effects of HDM, and long-term risk of developing secondary cancers.40,41 The development of melflufen for later-line treatment places melphalan in a more clinically and biologically rational space in the MM treatment paradigm, where patients have exhausted most of the available treatments. Although, initially, we believed that the later line use of this melphalan prodrug would have a favourable benefit to risk profile, the detrimental survival signal has led us to rethink this balance. Regardless, since the product has been withdrawn from the market, except for rare cases, it is not available for general use. In this practice, melflufen had reasonable tolerability, with toxicities being mostly haematological and treatable. Without experiencing increased deaths anecdotally, the OCEAN trial preliminary data and warning is strong enough for us to have terminated its use.
There are still several uncertainties regarding melflufen. First, is whether the current indication in a heavily pretreated population is the best use of melflufen, or whether there are synergistic combinations in less heavily treated populations that will prove better. Second, is regarding the suboptimal administration of melflufen, which currently requires central venous catheterization. The results of the PORT study are eagerly awaited in this context.38 Finally, it is unknown whether melflufen can be used in the high-dose setting with stem-cell rescue to replace traditional melphalan. Unfortunately, given the recent withdrawal of melflufen from the market, the above questions may never be answered as Oncopeptides has strategically changed the company’s direction. We are hopeful that in the near future, the void created will be filled by more tolerable and efficacious therapies as there is still a significant unmet need in RRMM. (https://www.oncopeptides.com/en/media/press-releases/oncopeptides-withdraws-pepaxto-in-us-scale-down-organization-and-focus-on-rd)