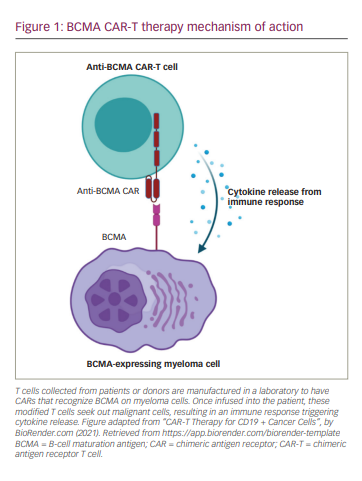
Background on anti-BCMA CAR-T therapy
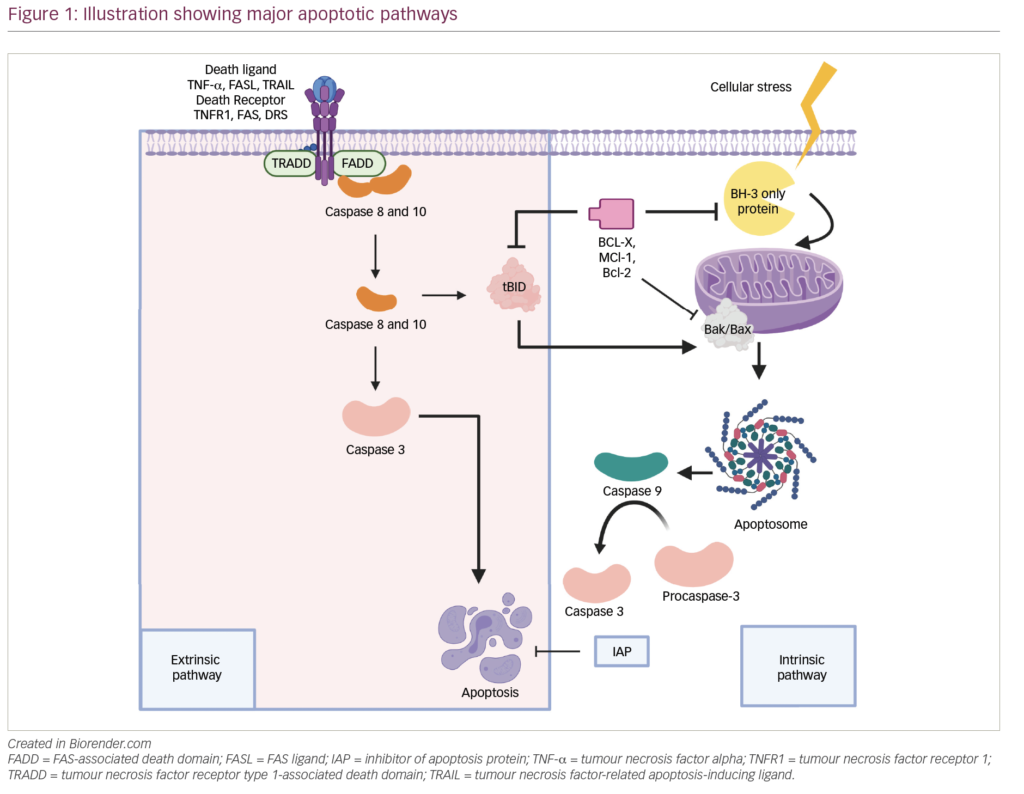
Despite notable advances in the development of new drugs and the improvement of survival rates over the last 20 years, multiple myeloma (MM) persists as an incurable disease and nearly 35,000 new cases are expected in the USA each year.1,2 The emergence of novel immunotherapies treatments has changed the therapeutic landscape for MM. One therapy of note, chimeric antigen receptor CAR T-cell (CAR-T) therapy, provides advanced specificity to T cells by manufacturing a patient’s own or donor T cells to recognize and attack malignant myeloma cells. This therapy combines antigen binding of a tumour cell-surface molecule and T-cell activation, initiating a targeted immune response (Figure 1). B-cell maturation antigen (BCMA) is currently the most prominent and promising antigen in MM and has shown impressive results in both response rate and depth of responses when used as the target for CAR-T therapy. Despite this, the majority of patients eventually relapse. The next step in evolving the treatment of MM is understanding the mechanism of response and relapse in patients treated with anti-BCMA CAR-T therapy. This knowledge will be essential to help guide the strategy of treatments post anti-BCMA therapy; be it sequential anti-BCMA therapies or utilizing new targets.
Anti-BCMA therapy use and failure
The development of various anti-BCMA immunotherapies has raised the question of retreatment to challenge the effect of a previous anti-BCMA therapy after relapse. In a study by Gazeau et al., the outcome of a patient who was retreated with an anti-BCMA therapy after a previous failure was assessed.3 The patient presented with kappa light-chain MM and was treated within a phase II trial of BCMA-directed CAR-T (Efficacy and safety study of bb2121 in subjects with relapsed and refractory multiple myeloma [KarMMa]).4 After having achieved a stringent complete response, the patient relapsed 1 year post CAR-T infusion. The study found that as this patient responded, soluble BCMA expression declined and then increased at relapse, suggesting malignant plasma cells (PCs) still expressed BCMA. The patient was retreated with the same CAR-T at the same dose following protocol, with no response. The persistent BCMA expression by tumour cells permitted the use of a different anti-BCMA therapy (belantamab mafodotin), which resulted in a very good partial response (VGPR) after three injections and continued response 5 months later (Table 1). This case suggests that patients whose disease has progressed after BCMA-targeted therapy might respond to alternate BCMA-targeted therapy; however, understanding the mechanism of their initial failure is imperative to subsequent treatment.
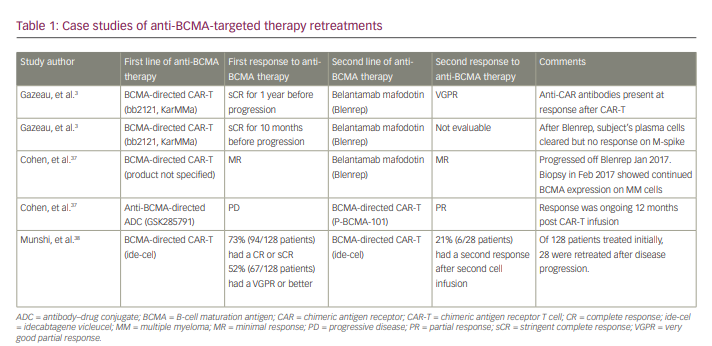
BCMA is an effective anti-myeloma target due to its specific surface expression on malignant PCs and its non-expression in normal tissue and haematopoietic cells. In normal B cells, BCMA regulates differentiation, survival and maturation. In the malignant PC, BCMA is associated with the cell’s uncontrolled proliferation and immunosuppressive bone marrow microenvironment.5 This antigen’s presence and specificity to malignant PCs thus makes it an ideal druggable target for anti-MM therapy, with minimal off-target toxicities in normal tissues.6 Overall, CAR-T therapies targeting BCMA have shown unprecedented and impressive improvements in response rates. The reported phase I–II data from the KarMMa-3 (ide-cel, bb2121), CARTITUDE-1 (cilta-cel, JNJ-68284528), orvacabtagene autoleucel (orva-cel; JCARH125) and LUMMICAR-2 (zevor-cel; CT053) studies show response rates ranging from 73% to 94.8% in heavily pre-treated patients.2,7 Table 2 provides information on current approved or pending approval anti-BCMA targeted therapies available for patients with MM, and updated bispecific BCMA/CD3-directed antibody response data can be found in Table 3. Despite the exceptional efficacy of these anti-BCMA-therapies, relapses have been observed in the majority of treated patients. The mechanisms for disease progression and therapeutic resistance remain largely in question, but antigen escape and T-cell exhaustion have been implicated as two likely culprits.8,9 Understanding these potential mechanisms can aid in physicians’ decisions to initiate subsequent treatment with either a regimen that re-challenges anti-BCMA therapy or one that utilizes another target entirely.

T-cell exhaustion
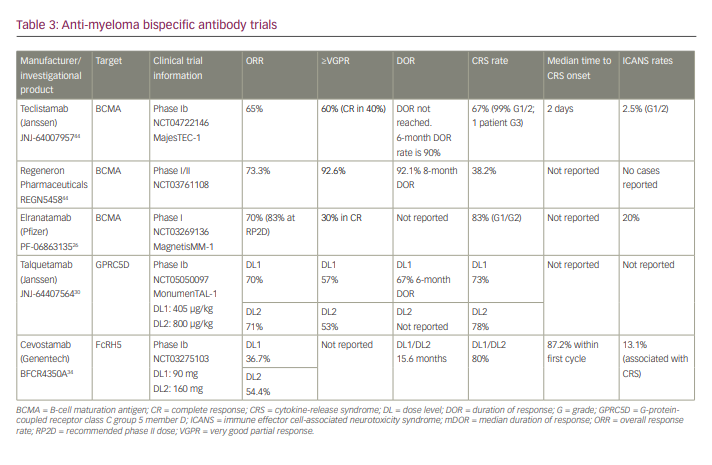
It is hypothesized that one of the main causes of CAR-T therapy failure and/or relapse is due to short-term T-cell persistence and poor T-cell expansion.10 This T-cell exhaustion results in effector T cells having reduced capacity to secrete cytokines, reduced proliferative activity and increased expression of inhibitory biomarkers.5 This exhaustion occurs in natural T cells in the case of prolonged antigen stimulation, often observed in instances of infection or cancer.11 In patients post CAR-T infusion, it can be reasonably speculated that these manufactured cells reach exhaustion after being constantly exposed to their antigen and needing to shut down to avoid causing too much collateral damage to the human body. The myeloma cells can then, in a sense, overburden the T cells to a point of breaking – and thus, exhaustion – therefore successfully resisting defeat. As previously noted, T-cell exhaustion is commonly marked in the increased expression of co-inhibitory receptors/pathways, notably programmed death receptor 1 (PD-1) and programmed death-ligand 1 (PD-L1) (Figure 2).12,13 One mechanism to bypass this exhaustion is to manufacture T cells that silence these inhibitors. A study by Penn Medicine investigated T-cell exhaustion by manufacturing a CAR-T-targeting mesothelin (a common marker in pancreatic tumours) and exposed the CAR-T to mesothelin-expressing pancreatic tumour cells for 4 weeks.14 T-cell exhaustion was accompanied by surges of proteins that acted as on–off switches of genes in immune cells. Translating this into MM therapy, regulation of these switches could bypass T-cell exhaustion and result in longer-lasting responses to anti-BCMA and other target CAR-Ts.
PD-1 is an inhibitory receptor expressed on T cells and enables the immune system to maintain tolerance to self-antigens. Cancer cells use this pathway to evade immune-mediated malignant cell death. Thus, PD-1 inhibitors reverse this immune suppression. There have been several clinical trials investigating the effect of immune checkpoint inhibition of PD-1 and PD-L1 in combination with CAR-T therapy in various haematologic malignancies.15 The data from these studies indicate that combining PD-1 blockade with CAR-T could result in exhausted T-cell reinvigoration. A retrospective study was completed with five patients who progressed through BCMA-targeted therapy and received PD-1 inhibitor (pembrolizumab) combination as their salvage therapy.16 All patients still had CAR-T BCMA cells detectable by real-time polymerase chain reaction prior to initiation of salvage therapy. One patient exhibited a minimal response at day 12, which was associated with expansion of CAR-T BCMA cells. This demonstrates that a PD-1 inhibitor combination has the potential to induce CAR-T re-expansion in patients progressing after CAR-T anti-BCMA therapy and may be an option to overcome this mechanism of resistance.
Antigen escape
One common limitation of CAR-T therapy is the development of tumour resistance to single antigen-targeting CARs, such as anti-BCMA CARs. After an initial promising response, malignant cells of patients treated with these CAR-T present with either partial or complete loss of target antigen expression (Figure 2).10 When this occurs it is called antigen escape and is expected when treatment targets are not essential to cellular function. Several studies have reported this phenomenon of downregulation/loss of BCMA expression in patients with MM treated with anti-BCMA CAR-T therapies after having achieved promising responses.17,18 These findings are consistent with the development of a mechanism of resistance by myeloma cells.
BCMA is crucial in regulating B-cell maturation and survival, as well as differentiation into PCs;19,20 therefore, loss of this antigen is expected. However, it is becoming increasingly more common in patients post anti-BCMA CAR-T treatment. One such case study of an immunoglobin A lambda MM patient enrolled in a phase I trial (CRB-401; ClinicalTrials.gov number NCT02658929) of anti-BCMA CAR-T therapy has been reported.21 This patient relapsed 9 months after the first cell infusion, and then received a second cell infusion with the same product at a higher dose with no response. Additional tissue studies revealed biallelic loss of BCMA, suggesting an antigen escape resistance mechanism. Thus, it can be interpreted that, while BCMA plays a critical role in PCs, malignant PCs may have the ability to reroute their growth mechanisms to overcome BCMA treatment regimens.
Anti-BCMA CAR-T and bispecific T-cell engager antibody treatments have begun to be explored in re-challenging patients treated with BCMA-targeted therapies utilizing gamma secretase inhibitors (GSIs).22 GS, which is found in PCs, is responsible for cleaning BCMA from the surface of PCs and releasing it into the blood as soluble BCMA. The rationale for combining GSI with BCMA-directed therapy is that GS-mediated cleavage of BCMA could limit the efficacy of BCMA-directed CAR-T by lowering target antigen expression; thus resulting in the antigen escape phenomenon. GSIs therefore reduce shedding of BCMA from MM cells, increase BCMA surface expression and enhance CAR-T recognition of MM cells.
A study by Cowan et al.23 investigated the use of anti-BCMA CAR-T therapy in combination with a GSI in patients with relapsing/remitting MM (RRMM). T cells were isolated via positive selection from patients with RRMM who showed ≥10% PCs in the bone marrow by CD138 immunohistochemistry and measurable disease by International Myeloma Working Group criteria. These T cells were then modified in vitro for BCMA-targeted CAR-T therapy. These patients received a GSI (JSMD194) for three (run-in) doses and a bone marrow aspirate was obtained to show BCMA expression on tumour cells compared with baseline. After lymphodepleting chemotherapy, BCMA CAR-T therapy was infused in combination with JSMD194 three times weekly for 3 weeks, starting on the day of CAR infusion. Among six assessable patients, the best overall response rate (ORR) was 100% (five VGPRs, one partial response). These results exhibit the potential efficacy of BCMA-targeted CARs in combination with GSI for patients with RRMM, paving the way for this combination in other BCMA-targeted immune effector cell regimens to overcome antigen escape.
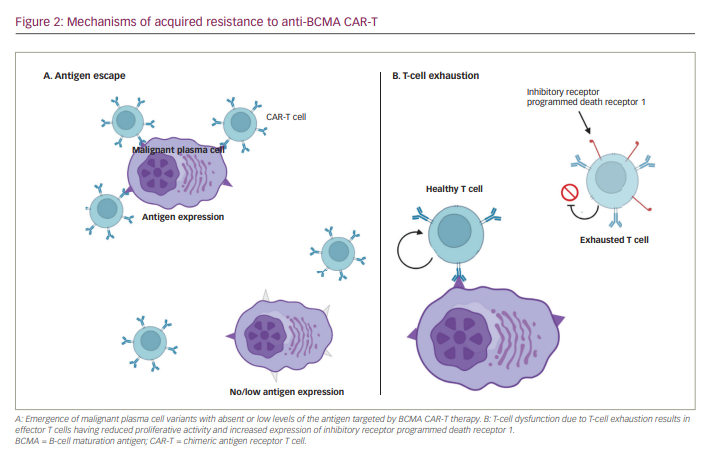
There is currently a trial open to enrollment that is investigating a bispecific T-cell-engaging antibody called teclistamab in combination with other anti-cancer therapies (ClinicalTrials.gov number NCT04722146). There are five regimens open on this trial, with four of them excluding patients with exposure to prior BCMA-targeting therapies. The only regimen that allows prior exposure is that which contains teclistamab and nirogacestat, a GSI. As noted previously, much of the clinical data supporting the hypothesis that GSI can help overcome mechanisms of resistance for BCMA-targeted therapies come from BCMA CAR-T studies. The teclistamab trial is utilizing this data to investigate whether the same positive results can occur when bispecific T-cell-engaging antibodies are paired with a GSI. It is one of few studies available currently that allows patients with prior exposure to BCMA-targeted therapy to be enrolled.
Re-challenging with BCMA-targeted therapies
Although transient downregulation or loss of BCMA expression has been seen following BCMA CAR-T therapy, most patients show persistent BCMA expression at progression.24 This finding suggests that patients may respond to subsequent BCMA-targeted treatment utilizing a different mechanism of action. As previously noted, there is currently a study of a bispecific T-cell-engaging antibody targeting BCMA (teclistamab) that is allowing subjects with previous exposure to anti-BCMA therapy to be enrolled onto a regimen including a GSI, nirogacestat. Another study demonstrated that a patient who progressed through BCMA-targeted CAR-T therapy responded to treatment with belantamab mafodotin, an antibody–drug conjugate.3 Additionally, a recent phase I trial of elranatamab, a BCMA/CD3 bispecific antibody, did include four patients treated with previous BCMA-targeted therapy. Three of those patients achieved a response, with one stringent complete response and two VGPRs.25,26
One possible BCMA-targeted re-challenging treatment could include the use of daratumumab. Daratumumab is a human monoclonal antibody that binds CD38-expressing PCs, thus inducing tumour cell death through antibody-dependent cell-mediated cytotoxicity, antibody-dependent phagocytosis, apoptosis and complement-dependent cytotoxicity.27 It is hypothesized that the combination of anti-BCMA immune effector cell therapies with daratumumab will lead to a synergistic anti-myeloma effect. The potential benefits of combining these therapies are thought to be twofold: Daratumumab has direct cytotoxicity to myeloma cells, and can lead to increased T-cell-mediated killing of MM cells by BCMA-targeted therapies due to daratumumab’s ability to increase cytotoxic helper T cells and deplete CD38+ immunoregulatory cells.
The development of various anti-BCMA immunotherapies has resulted in exploring sequential anti-BCMA therapies as a treatment option to re-challenge after relapse. Therapies with different mechanisms of action include bispecific T-cell-engaging antibodies, antibody–drug conjugates and tandem dual CAR-T therapies, all of which are becoming increasingly prevalent in MM clinical trials. However, most trials exploring BCMA-targeted therapies exclude patients with prior anti-BCMA treatments. This is hindering both the treatment options for these relapsed patients, and the clinical trial data on sequential treatment with BCMA-targeted therapies. While these options are limited for patients, it is critical that other novel targets are explored as treatment options for patients with RRMM.
New targets
MM has several surface molecules and molecular pathways that are druggable targets, such as CD38, G-protein-coupled receptor class C group 5 member D (GPRC5D), signalling lymphocytic-activation molecule (SLAM) family member 7 (SLAMF7), and Fc receptor-homolog 5 (FcRH5). The variability of surface receptors on myeloma cells provides new avenues for immune effector therapies in patients with anti-BCMA therapy-refractory MM. Many of these novel treatments manipulate the body’s own immune system to target cancer. Cancer’s natural ability to evade the immune system has proved to be the largest obstacle for oncologists for several centuries. However, the development of manufactured T cells has provided patients with a frontline immune system that is not as easily deceived by the elusive nature of malignancy.
GPRC5D
GPRC5D is a therapeutically targeted type-C7 transmembrane protein expressed on the surface of CD138+ malignant PCs.27,28 Levels of GPRC5D mRNA are elevated in patients with myeloma phenotypes and are correlated with high-risk PC malignancies, making this a promising target for T-cell-mediated killing.29 Though both BCMA and GPRC5D surface receptors are present in a majority of MM cell lines, GPRC5D expression is heterogeneous and independent of BCMA expression. Using CRISPR technology, it has been shown that BCMA knockout does not affect the cytotoxicity and expression of GPRC5D-expressing cells.29 In cases of antigen escape or BCMA downregulation in patients previously exposed to BCMA-targeted therapy, these data suggest that loss of BCMA will not affect response rates in CD138+ and GPRC5D-expressing phenotypes.
Updated phase I results from the MonumenTAL-1 study have been reported.30 In total, 95 patients, all with triple-class-exposed disease, received either 405 μg/kg weekly or 800 μg/kg biweekly subcutaneous dosing. In the weekly dosing cohort, 17% of patients had previously received BCMA-directed therapy. Responses deepened over time and were durable, with ORRs of 70% and 71%, respectively. Median duration of response was not met in either of dosing cohorts and the 6-month duration of response rate in the weekly dosing cohort was 67%. Pharmacodynamic data between cohorts were similar. The cytokine-release syndrome rate in the weekly dosing group was 73% (all grade 1/2, except one grade 3) and 78% (all grade 1/2) in the biweekly dosing group. Additional off-tumour toxicities were observed, compounding effects to receptors present on the skin, hair follicles and mouth.28,31 Skin-related adverse events were seen in 77% – all grade 1/2 – and nail changes were seen in 30% of patients.
In an effort to combine anti-MM cytotoxicity and decrease suppressive CD38+ immunoregulatory cells, 23 patients with RRMM received subcutaneous talquetamab in combination with daratumumab in a phase Ib multi-cohort study (TRIMM-2; ClinicalTrials.gov NCT0410819532). Cohorts included daratumumab 1,800 mg in combination with talquetamab 400 μg/kg weekly (n=8), talquetamab 400 μg/kg biweekly (n=5) or talquetamab 800 μg/kg weekly (n=9). Patients had received an average of six lines of prior therapy and 73.9% were penta-exposed. The rate of cytokine-release syndrome across cohorts was 55.2% (all grades 1/2), with a median onset of 2.5 days. Dysgeusia was common at 48.3% (all grade 1/2), and skin toxicity occurred in 27.6%. Cases of both grade 1 and grade 3 immune effector cell-associated neurotoxicity syndrome were also reported. ORRs in each cohort were 80%, 85.7% and 77.8%, respectively, and deepened over time. Correlative studies showed peripheral T-cell activation, with upregulation of CD38+/CD3+ cells.
FcRH5
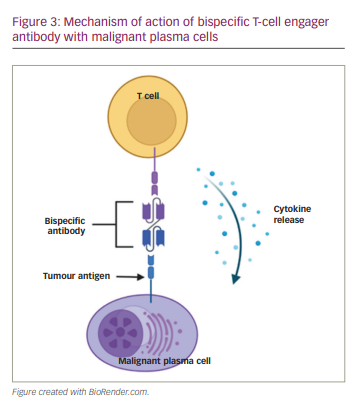
FcRH5 is a type I membrane protein expressed on B cells, PCs, and, most importantly, myeloma cells, with nearly 100% prevalence.33 Cevostamab (BFCR4350A) is an anti-FcRH5/CD3 humanized immunoglobulin that initiates dual binding to myeloma FcRH5 cell surface markers and the CD3 T-cell protein complex.33 This bispecific engagement creates a bridge to allow close-proximity immunogenicity between the immune system and myeloma cells (Figure 3). A phase I trial (NCT03275103) is being conducted in high-risk patients with a median of six prior lines of therapy and exposure to several drug classes, including previously targeted BCMA therapy. Data have been reported on 160 patients, of whom 85% had
triple-class-refractory disease.34 At a median 6-month follow up, the median time to response was 29 days. Responses appeared to be dose dependent, with an ORR of 54.4% at the higher dose level of 160 mg compared with 36.7% at the 90 mg dose level. With target dose levels greater than 90 mg, patients with prior anti-BCMA therapy (n=8/22) had an ORR of 36.4%. Estimated duration of response for all responders was 15.6 months. The results of this trial are promising for diminishing the disease burden of many RRMM patients.
SLAMF7
SLAMs are transmembrane receptors capable of regulating immune cell function.35 This modulatory function is accomplished by an interaction between tyrosine-based mechanisms and intracellular adapter proteins.35 SLAMF7 is a member of this transmembrane receptor class and is highly expressed in RRMM patients. This receptor functions to conciliate either inhibitory or activating factors among immune cells through intracellular signalling. Though this mechanism is not entirely understood, it is believed that these processes are involved in myeloma cell survival. SLAMF7 receptors are present on a variety of immune cells such as natural killer cells, subsets of T cells, monocytes, macrophages and dendritic cells. Due to its lack of presence on any other known human tissue, this target is an ideal candidate for CAR-T-targeted therapies. Several trials are currently in place to study the anti-myeloma effects and maximum tolerated doses of SLAMF7-targeted CAR-T products. In a phase I/IIa trial, RRMM patients who have exhausted conventional therapies are being given an autologous SLAMF7 CAR-T product to evaluate the anti-tumor ability and safety of the immune effector product (CARAMBA-1; ClinicalTrials.gov NCT04499339). Similarly, a phase I study of the CAR-T cell biologic UCARTCS1A is exploring the maximum tolerated dose for anti-myeloma effects in RRMM patients (ClinicalTrials.gov NCT04142619).
CD38
Previously explored anti-CD38 therapies have included the monoclonal antibodies daratumumab and isatuximab. However, most patients will progress through this treatment due to resistance mechanisms and persistent malignancy.36 Recent therapeutic approaches have attempted to utilize this same target for CARs and bispecifics antibodies. In a preclinical trial studying the efficacy of Bi38-3, a CD38/CD3 bispecific T-cell engager, in vitro, ex vivo and in vivo results indicated that this immune effector cell product can direct T-cell activity towards myeloma cells expressing CD38.36 Dependent on the dose of Bi38-3, cytotoxicity of the CD138+ MM cells was apparent in samples of both recently diagnosed and relapsed tumour cells.36 Additionally, reduced toxicity of peripheral blood mononuclear cells (T, B and natural killer cells) and CD34+ haematopoietic bone marrow was observed.36 There was no significant cell death among these cells, showing >40% survival rate. In general, CD38/CD3-mediated cytotoxicity was abundantly prevalent in cells expressing high levels of CD38, and there was limited toxicity among those with low-to-intermediate levels of CD38 expression, such as the body’s own haematopoietic progenitors.36 The data covering differing levels of toxicity in malignant PCs versus non-malignant cells outline the importance of not only reducing disease burden but preserving the body’s own immune cells. With this approach, we are able to heighten the immune system’s natural defense with pharmaceutical intervention.
Conclusion
The use of immune effector cell therapies in treating MM has resulted in remarkable progress in the field of oncology. With their relatively high response rates and chances of remission, these therapies have become the frontline of evolution into treating this incurable disease. With anti-BCMA CAR-T therapy heading the revolution of novel therapies, the daunting question remains: What next? With the inevitable relapse of patients fighting this disease, where do we go after exhausting the most promising route of therapy? Mechanisms of resistance mediate this inevitable relapse in patients, giving rise to the continual need for more treatment options. Antigen escape and T-cell exhaustion are just two examples of the elusive and strenuous impact that malignancy has on the immune system. BCMA downregulation may occur in patients previously treated with anti-BCMA CAR-T cells, diluting the potential for BCMA re-challenge. Similarly, overly exerted T cells may demonstrate diminished anti-myeloma activity and cytokine release when exposed to a target antigen. When faced with these perpetrators of relapse, the treatment options for an anti-BCMA therapy refractory patient then come down to two: Re-challenging BCMA or directing the immune system at a novel target. In the case of re-challenging BCMA, this can be efficacious when enacting a new mechanism of action. Paired with GSIs or monoclonal antibodies, responses can again be seen in patients with RRMM who have previously been exposed to anti-BCMA therapy. When re-challenging is not an option, therapy against a novel target can prove effective against relapsing myeloma. As new mechanisms of immune effector therapies are paired with the discovery of new targets, we are seeing new avenues into the next frontier of MM treatment.