Endocrine therapy (ET) has changed the natural history of hormone receptor-positive (HR+) breast cancer (BC) and is the cornerstone of the treatment of HR+ BC. There are several ETs approved for the treatment of BC, including selective oestrogen receptor modulators (SERMs; tamoxifen), aromatase inhibitors (AIs; anastrazole, letrozole and exemestane) and selective oestrogen receptor degraders (SERDs; fulvestrant and elacestrant).1–3 Additionally, several targeted agents have been approved, including cyclin-dependent kinase 4/6 inhibitors (CDK4/6i), protein kinase B (AKT) inhibitors, phosphoinositide 3-kinase (PIK3CA) and mammalian target of rapamycin (mTOR) inhibitors, which have been shown to improve patient outcomes when given in combination with ET.4–9
When progression of disease (PD) occurs during ET, endocrine resistance is often implicated. Primary endocrine resistance is defined as either a relapse during the first 2 years of adjuvant ET or PD within the first 6 months of first-line ET for advanced breast cancer (aBC). Secondary endocrine resistance is defined as a relapse after the first 2 years of endocrine therapy, a relapse within 12 months of completing adjuvant ET, or PD occurring 6 months after initiating ET for aBC (Figure 1A).10 There is a need to understand the mechanisms of endocrine resistance to develop novel agents, optimize treatment selection and sequencing, and ultimately improve patient outcomes in the aBC setting.12 This article highlights the landscape of pharmacological and clinical trial data regarding novel agents for the treatment of HR+/human epidermal growth factor 2-negative (HER2-) aBC.
Figure 1: Clinical definitions of endocrine resistance
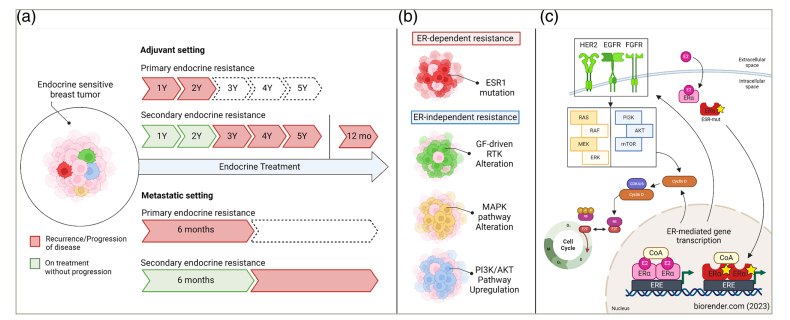
Created with biorender.com (2023).
(a) Clinical definitions of endocrine resistance based on the European School of Oncology–European Society of Medical Oncology international consensus guidelines for advanced breast cancer.10 Pathways (b) and primary mechanisms (c) being investigated for potential clinical application in addressing oestrogen receptor-dependent and oestrogen receptor-independent endocrine resistance.
Primary endocrine resistance is defined as a relapse during the first 2 years of adjuvant ET or the progression of disease within the first 6 months of first-line ET for advanced breast cancer. Secondary endocrine resistance is defined as a relapse during adjuvant ET after the first 2 years, relapse within 12 months of completing adjuvant ET, or the progression of disease ≥6 months after initiating ET for advanced breast cancer while receiving ET. Although ER-dependent and ER-independent pathways are presented separately for conceptual clarity, significant intracellular crosstalk exists between these pathways. The green, yellow and blue factors participate in ER crosstalk, and disruptions in mitogenic signalling pathway components can contribute to tumour growth independent of ER and resistance to selective oestrogen receptor degraders.
AKT = protein kinase B; CDK = cyclin-dependent kinase; CoA = coactivator; EGFR = epidermal growth factor receptor; ER = oestrogen receptor; ERE = oestrogen response element; ERK = extracellular signal-regulated kinase; ESR1 = oestrogen receptor 1; ESR1-mut = ESR1 mutant; ET = endocrine therapy; FGFR = fibroblast growth factor receptor; GF = growth factor; HER2 = human epidermal growth factor receptor 2; MAPK = mitogen-activated protein kinase; MEK = meiotic chromosome-axis-associated kinase; mo = months; mTOR = mammalian target of rapamycin; p = phosphate; PI3K = phosphoinositide 3-kinase; RAS = rat sarcoma; RAF = rapidly accelerated fibrosarcoma; RB = retinoblastoma; RTK = receptor tyrosine kinase; Y = year.10
Figure 1C: Adapted from Lloyd et al., 2022.11 (https://creativecommons.org/licenses/by-nc/4.0/).
Mechanisms of endocrine resistance
The oestrogen receptor (ER) is a ligand-dependent transcription factor. Once oestrogen binds to ER, it dimerizes and associates with DNA at the oestrogen response element (ERE), which then mediates gene transcription and promotion of cell proliferation.12,13 The ER also leads to non-genomic signalling by directly stimulating receptor tyrosine kinases (RTKs), such as HER2, insulin-like growth factor 1 (IGF-1) and epidermal growth factor receptor (EGFR), to activate mitogen-activated protein kinase (MAPK) and PI3K/AKT oncogenic signal transduction pathways, as shown in Figure 2.14
Figure 2: Mechanism of action of oestrogen receptor inhibitors used in breast cancer treatment
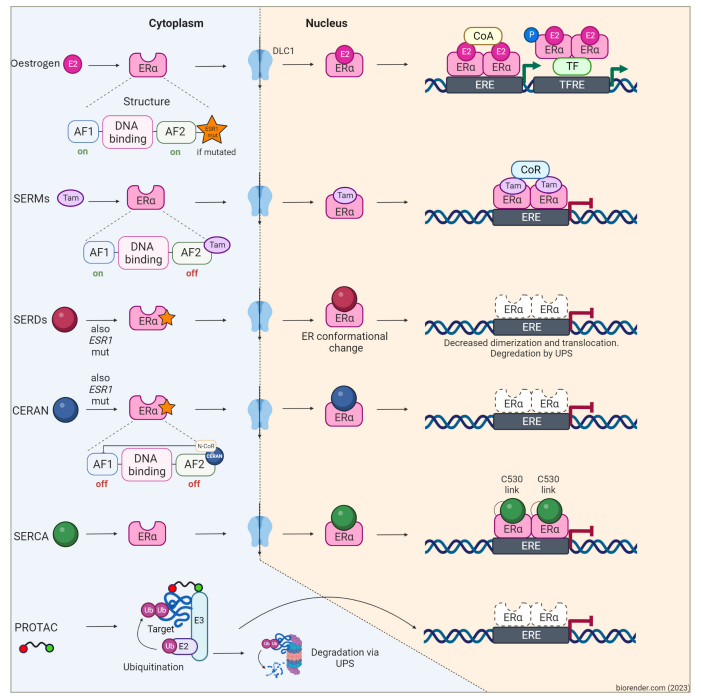
Figure created with biorender.com (2023).
In oestrogen-sensitive cells, oestrogen binds to its ER. This binding results in the formation of oestrogen–ER complexes, which then dimerize and translocate into the nucleus. Subsequently, these complexes bind to EREs found in multiple genes, recruiting ER coactivator proteins and enhancing target gene transcription. This process ultimately promotes cancer cell proliferation and survival. SERMs, such as tamoxifen, bazedoxifene and lasofoxifene, act as antagonists of gene transcription in breast cancer cells but function as agonists in other tissues upon binding to the ER. Among SERDs, fulvestrant stands as the most significant representative. It operates by inhibiting ER dimerization and translocation into the nucleus while facilitating proteasomal degradation. Fulvestrant demonstrates partial activity even in ESR1-mutant ER, which would otherwise facilitate gene transcription independent of the presence of the ligand. Oral SERDs, CERANs and SERCAs ultimately achieve effects similar to fulvestrant; however, they exhibit a more potent activity on both wild-type and mutant ER, leading to a higher rate of receptor degradation. Lastly, PROTACs comprise a domain binding to a target protein and another domain binding to an E3 ubiquitin ligase. The proximity of these elements enhances the target protein’s susceptibility to polyubiquitination and subsequent proteasomal degradation of ER in cancer cells.
AF1 = activation function 1; AF2 = activation function 2; CERAN = complete oestrogen receptor antagonist; CoA = steroid receptor coactivator; CoR = steroid receptor corepressor; DLC1 = deleted in liver cancer 1; E2 (pink) = oestradiol; E3 = ubiquitin–protein ligase; ERα = oestrogen receptor alpha; ERE = oestrogen response elements; ESR1 = oestrogen receptor 1; E2 (purple) = ubiquitin-conjugating enzyme; P = phosphorus; PROTAC = proteolysis-targeting chimera; SERCA = selective oestrogen receptor covalent antagonist; SERD = selective oestrogen receptor degrader; SERM = selective oestrogen receptor modulator; Tam = tamoxifen; TF = transcription factor; TFRE = transcription factor response element; Ub = ubiquitin; UPS = ubiquitin–proteasome system.
Oestrogen receptor-dependent mechanisms of resistance
Mutations in the ligand-binding domain of oestrogen receptor 1 (ESR1) promote ET resistance through hormone-independent ER signalling, upregulated coactivator binding and stability against proteolytic degradation (Figures 1B and 2).11 ESR1 mutations are acquired in 25–40% of tumours of patients pretreated with ET.15,16 ESR1 mutations stabilize the ER in the active conformation, which enables the binding of coactivators in the absence of ligand, leading to AI resistance and decreased sensitivity to tamoxifen and fulvestrant.17 Downregulation of the ER is another potential cause of ET resistance.16 Approximately 20% of ER+ BCs lose ER over time with ET.18
Selected oestrogen receptor-independent mechanisms of resistance
Some tumours lose sensitivity to ET by the activation of other pathways such as membrane receptor pathways, upregulation of oncogenic transduction and dysregulation of the cell cycle through the CDKs, hence the rationale for combining ET with other targeted drugs (Figure 1C).19 RTKs are a family of membrane proteins that contain an intracellular tyrosine kinase component. Mutations or amplifications of RTKs lead to the initiation of intracellular signal transduction along the MAPK and PI3K/AKT pathways, which activate transcriptional activity of ER in the absence of oestrogen signalling.20–24
The PI3K/AKT pathway is critical to cell growth and survival.25 Aberrant activation of the PI3K pathway promotes the acquired resistance to oestrogen depletion in preclinical models.26
PI3K is a membrane-bound enzyme activated by RTK and G-protein-coupled receptors. AKT (a serine/threonine kinase) is the principal downstream molecule of the PI3K pathway.27 Activated AKT mediates the regulation of the cell cycle, growth and proliferation.28 mTOR is a downstream effector in the PI3K/AKT pathway.29 Phosphatase and tensin homologue (PTEN) acts as a tumour suppressor, which blocks AKT phosphorylation in the PI3K/AKT/mTOR pathway and inhibits cell proliferation. Loss of PTEN has been found to be associated with increased cell proliferation and survival.27 Mutations in genes encoding for components of the PI3K/AKT pathway occur frequently in BCs; about 30–40% of patients with HR+/HER2- BC have an activating mutation of the catalytic (p110) subunit of PI3K.27 As alterations in this pathway are common in BC, it is an attractive target for treatment.
Cyclins and CDKs help to regulate the cell cycle and gene transcription. In humans, there are 20 CDKs and 29 cyclins.30 CDK4/6 and their partner D-type cyclins (cyclins D1, D2 and D3) regulate the transition from G1 to S phases of the cell cycle.31 The cyclin D–CDK4/6 complex then binds p21 or p27 and phosphorylates retinoblastoma (Rb) protein. Phosphorylated Rb induces partial de-repression of E2F transcription factors and expression of cyclin E genes. Cyclin E then partners with CDK2 to hyper-phosphorylate Rb and establish progression to the S phase.31–33 While CDK 4/6 inhibition has shown improved progression-free survival (PFS) and overall survival (OS), the activation of CDK2/cyclin E is one resistance mechanism by which tumour cells can develop CDK4/6 resistance.4–6,34 The gene CCNE1 encodes cyclin E1, and it is downstream of the cyclin D1–CDK 4/6 pathway. Cyclin E1 can bind CDK2 and phosphorylate Rb independently, leading to the progression of the cell cycle, bypassing CDK 4/6.35 CDK7 is a part of the CDK-activating kinase (CAK) responsible for phosphorylating other cell cycle CDKs (CDKs 1, 2, 4 and 6) and driving progression through the cell cycle. CAK also complexes with the core human transcription factor II basal transcription complex and mediates gene transcription by activating RNA polymerase II.36–38 CDK7 also modulates ER activity through serine 118 phosphorylation.39
Novel endocrine therapies
Selective oestrogen receptor degrader
While many BCs develop endocrine resistance after treatment with ET, those that do so through ESR1 mutations still rely on ER-mediated signalling for growth, making the ER still a viable target for treatment (Figure 2). SERDs increase ER degradation, slow ER nuclear translocation and reduce transcription of ER-regulated genes.3,11 Fulvestrant was the first SERD to be approved for the treatment of metastatic HR+ BC.40–43 Some limitations of this agent include the route of administration (intramuscular) and the somewhat modest clinical activity as monotherapy after progression on AI and CDK4/6i, with most patients experiencing progression within 3 months or less.44
Oral SERDs have been an area of active research in the past few years, with the aim of improving the ease of administration and activity in this class of agents. Elacestrant is an oral SERM/SERD hybrid drug and is the first oral SERD approved based on the results from the phase III EMERALD trial (Elacestrant Monotherapy vs. Standard of Care for the Treatment of Patients With ER+/HER2- Advanced Breast Cancer Following CDK4/6 Inhibitor Therapy: A Phase 3 Randomized, Open-label, Active-controlled, Multicenter Trial; ClinicalTrials.gov identifier: NCT03778931).45 This study enrolled 477 patients with HR+/HER2- aBC previously treated with ET, CDK4/6i and up to one line of chemotherapy for advanced disease. They were randomized to receive elacestrant versus standard ET (fulvestrant or AI) and stratified by ESR1 mutation status. In the intention-to-treat population, 12-month PFS was 22.3% in the elacestrant arm and 9.4% in the control arm (hazard ratio [HR] 0.70; 95% confidence interval [CI] 0.55–0.88; p=0.0018). In the ESR1-mutant patients, there was greater benefit with 12-month PFS of 26.6 versus 8.2% in the ESR1-wild-type (WT) group (HR 0.55; 95% CI 0.39–0.77; p=0.0005). The absolute PFS benefit was modest in the overall population and in those with ESR1 mutations (Table 1).8,45–51,53–55 The most common adverse events (AEs) were gastrointestinal side effects (nausea and vomiting) and fatigue.45 Longer duration of prior CDK4/6i in the metastatic setting (n=465) was found to be associated with longer PFS on elacestrant versus standard-of-care (SoC) ET. In patients with an ESR1 mutation, the median PFS (mPFS) for those who received prior CDK4/6i for at least 12 months was 8.6 versus 1.9 months (HR 0.41, 95% CI 0.26–0.63) in the elacestrant arm (n=78) versus control arm (n=81), respectively.60 In January 2023, elacestrant was approved by the US Food and Drug Administration (FDA) for patients with HR+/HER2- aBC with ESR1 mutations after the progression on at least one line of ET.61
Table 1: Selected published clinical trials assessing novel endocrine therapies in hormone receptor-positive breast cancers8,45–55
| Agent/class | Clinical trial identifier/phase | Trial description/sample size | Mutation status | Prior fulvestrant | Prior CDK4/6i | PFS endpoint (months) | Endpoint met? |
| Elacestrant/oral SERD | NCT03778931 (EMERALD)45/phase III | E: elacestrant C: SoC ET in HR+ aBC progressed on prior ET + CDK4/6i N=477 | ESR1m: 48% (E) and 47.2% (C) | Allowed (29.3% E versus 31.5% C) | Required (100%) | Overall: 2.8 (E) versus 1.9 (C) ESR1m: 3.8 (E) versus 1.9 (C) | Yes in ESR1m |
| Giredestrant/oral SERD | NCT04576455 (aceIERA)47,56/phase II | E: giredestrant C: physician-choice ET in HR+/HER2- aBC N=303 | ESR1m: 44% (E) and 34% (C) | Allowed (20% E versus 18% C) | Allowed (43% in E versus 41% in C) | 5.6 (E) versus 5.4 (C) HR 0.81; 95% CI 0.6–1.1; p=0.18 | No |
| Camizestrant/oral SERD | NCT04214288 (SERENA-2)46/phase II | E: camizestrant (75 mg [C75] and 150 mg [C150]) C: fulvestrant in HR+/HER2- aBC N=240 | ESR1m: 36.7% of total | Not allowed | Allowed (49.6% of total) | Overall: 7.2 (C75, HR 0.58; 90% CI 0.41–0.81; p=0.0124), 7.7 (C150, HR 0.67; 90% CI 0.48–0.92; p=0.0161), 3.7 (C) ESR1m: 6.3 (C75, HR 0.33; 90% CI 0.18–0.58), 9.2 (C150, 90% CI 0.55; 0.33–0.89) and 2.2 (C) | Yes |
| Amcenestrant/oral SERD
| NCT04059484 (AMEERA-3)48/phase II | E: amcenestrant C: ET in ER+/HER2- aBC with prior exposure to ET N=290 | ESR1m: 46.4% (E) and 39.3% (C) | Allowed (9.8% E versus 9.5% C) | Allowed (79.7% E versus 78.2% C) | 3.6 (E) versus 3.7 (control) HR 1.051; 95% CI 0.789–1.4; one-sided p=0.6437 | No |
| NCT04478266 (AMEERA-5)53,57/phase III | E: amcenestrant + palbociclib C: letrozole + palbociclib in treatment-naive HR+/HER2- aBC N=1,068 | Not reported | Not allowed | Not allowed | 14.1 (E) versus 16.6 (C) HR 1.209; 95% CI 0.94–1.56; p=0.93 | No | |
| Lasofoxifene/next-generation SERM | NCT03781063 (ELAINE-I)58/phase II | E: lasofoxifene C: fulvestrant in HR+/HER2- aBC with prior AI + CDK4/6i N=103 | ESR1m: required (100%) | Not allowed | Required (100%) | 6.04 (E) versus 4.04 (C) HR 0.699; 95% CI 0.445–1.125; p=0.138 | No |
| Alpelisib/PI3K inhibitor | NCT02437318 (SOLAR-1)8/phase III | E: alpelisib + fulvestrant C: placebo + fulvestrant in HR+/HER- aBC treated with prior AI N=572 | PIK3CAm: 29.5% (E) and 30% (C) | Not allowed | Allowed (6.1% of total) | No PIK3CAm: 7.4 (E) versus 5.6 (C) HR 0.85; 95% CI 0.58–1.25 PIK3CAm: 11 (E) versus 5.7 (C) HR 0.65; 95% CI 0.50–0.85; p<0.001 | Yes in PIK3CAm |
| NCT03056755 (BYLieve)49/phase II | Cohort A: alpelisib + fulvestrant prior AI + CDK4/6i Cohort B: alpelisib + letrozole prior fulvestrant + CDK4/6i Cohort C: alpelisib + fulvestrant prior chemo/ET in HR+/HER2- PIK3CAm aBC N=379 (cohort A: n=127, cohort B: n=126 and cohort C: n=126) | PIK3CAm: required (100%) | Patients in cohort B | All patients in cohorts A + B | Cohort A: 8.0 Cohort B: 5.6 Cohort C: 5.6 | Yes | |
| Inavolisib/PI3K inhibitor | NCT04191499 (INAVO120)50/phase III | E: inavolisib + fulvestrant + palbociclib C: placebo + fulvestrant + palbociclib in HR+/HER- aBC, progressed during/within 12 months of adjuvant ET completion, with no prior treatment for aBC N=325 | PIK3CAm: required (100%) | Not allowed | Not allowed | 15 (E) versus 7.3 (C) HR 0.42; 95% CI 0.32–0.59; p<0.0001 | Yes |
| Capivasertib/AKT inhibitor | NCT01992952 (FAKTION)59 /phase II | E: fulvestrant + capivasertib C: fulvestrant + placebo versus in HR+/HER2- aBC with prior AI N=140 | PI3K/AKT/PTEN pathway mutation: 51% (E) and 47% (C) | Not allowed | Not specified | Overall: 10.3 (E) versus 4.8 (C) HR 0.56; 95% CI 0.38–0.81; p=0.0023 PI3K/AKT/PTEN pathway altered: 12.8 m (E) versus 4.6 m (C) HR 0.44; 95% CI 0.26–0.72; p=0.0014 | Yes |
| NCT04305496 (CAPItello-291)51/phase III | E: capivasertib + fulvestrant C: placebo + fulvestrant in HR+/HER2- aBC progressed on AI N=708 | AKT pathway alteration: 40.8% of total | Not allowed | 69.1% of total | Overall: 7.2 (E) versus 3.6 (C) HR 0.60; 95% CI 0.51–0.71; p<0.001 AKTm: 7.3 (E) versus 3.1 (C) HR 0.50; 95% CI 0.38–0.65; p<0.001 | Yes in AKT pathway altered | |
| Ipatasertib/AKT inhibitor | NCT03337724 (IPATunity130)55/phase III | Cohort B E: ipatasertib + paclitaxel C: placebo + paclitaxel in PIK3CA/AKT1/ PTEN-altered HR+/HER2- aBC without prior ET N=222 | PIK3CA/AKT1/PTEN mutation required (100%) | Not allowed | Not allowed | 9.3 (E) versus 9.3 (C) HR 1.00; 95% CI 0.71–1.40; p=1.00 | No |
aBC = advanced breast cancer; AI = aromatase inhibitor; AKT = protein kinase B; AKTm = protein kinase B pathway alteration; C = control; C75 = camizestrant 75mg; C150 = camizestrant 150mg; CDK4/6i = cyclin-dependent kinase 4/6 inhibitor; CI = confidence interval; E = experimental; ESR1m = oestrogen receptor 1 mutant; ET = endocrine therapy; HER2- = human epidermal growth factor negative; HR+ = hormone receptor positive; HR = hazard ratio; PFS = progression-free survival; PI3K = phosphoinositide 3-kinase; PIK3CAm = phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha mutant; PTEN = phosphatase and tensin homologue; SERD = selective oestrogen receptor degrader; SERM = selective oestrogen receptor modulators; SoC = standard of care.
Camizestrant is another oral SERD, which has shown PFS benefit over fulvestrant based on the phase II SERENA-2 trial (A Randomised, Open-Label, Parallel-Group, Multicentre Phase 2 Study Comparing the Efficacy and Safety of Oral AZD9833 Versus Fulvestrant in Women With Advanced ER-Positive HER2-Negative Breast Cancer [SERENA-2]; ClinicalTrials.gov identifier: NCT04214288).46 Results are summarized in Table 1. SERENA-2 included 240 patients with HR+/HER2- aBC who had progressed on one line or less of prior ET and were randomized to receive camizestrant (75 or 150 mg) or fulvestrant. Of the enrolled patients, at baseline, 36.7% had detectable ESR1 mutation and 58.3% had lung/liver metastases. Patients treated with camizestrant at either dose had longer PFS as compared with those treated with fulvestrant. This difference was seen in tumours with and without an ESR1 mutation. Notable AEs likely related to camizestrant included grade 1 to 2 photopsia (18.4%) and sinus bradycardia (13.6%).46 The SERENA-1 phase I trial (A Phase 1 Dose Escalation and Expansion Study of AZD9833 Alone or in Combination in Women With ER-positive, HER2-negative Advanced Breast Cancer [SERENA-1]; ClinicalTrials.gov identifier: NCT03616587) is also evaluating camizestrant in combination with various other drugs, such as CDK 4/6, PI3K and mTOR inhibitors, in patients with HR+/HER2- aBC, with data available.62–64
Giredestrant is another oral SERD under development and was well tolerated in a phase I study (A Phase Ia/Ib, Multicenter, Open-Label, Dose Escalation, Dose Expansion Study Evaluating the Safety, Pharmacokinetics, and Activity of GDC-9545 Alone or in Combination With Palbociclib and/or LHRH Agonist in Patients With Locally Advanced or Metastatic Estrogen Receptor-Positive Breast Cancer; ClinicalTrials.gov identifier: NCT03332797).65 Giredestrant was compared with physician-choice ET in the phase II acelERA trial (A Phase II, Randomized, Open-Label, Multicenter Study Evaluating the Efficacy and Safety of GDC-9545 Compared With Physician’s Choice of Endocrine Monotherapy in Patients With Previously Treated Estrogen Receptor-Positive, HER2-Negative Locally Advanced or Metastatic Breast Cancer; ClinicalTrials.gov identifier: NCT04576455) as second- and third-line treatment in patients with HR+/HER2- aBC. However, the study did not reach significance for the primary endpoint of investigator-assessed PFS (Table 1).47,56 An exploratory subgroup analysis revealed the most pronounced benefit in the subgroup with ESR1 mutation.66 In that subgroup, PFS was 5.3 months in the giredestrant arm versus 3.5 months in the control arm (HR 0.53; 95% CI 0.33–0.93). Clinical benefit rate (CBR) was 25.5 versus 2.6%, and objective response rate (ORR) was 13.7 versus 0% in the giredestrant versus control arms, respectively. Girdestrant is well tolerated and AEs are comparable to those of ET. AE leading to treatment discontinuation occurred in 1 versus 2% of the giredestrant versus control arms, respectively.66 These secondary outcomes have prompted continued investigation of giredestrant in the HR+/HER2- aBC setting.
Amcenestrant is an oral SERD that was tested in the phase III AMEERA-5 trial (A Randomized, Multicenter, Double-blind Phase 3 Study of Amcenestrant [SAR439859] Plus Palbociclib Versus Letrozole Plus Palbociclib for the Treatment of Patients With ER [+], HER2 [-] Breast Cancer Who Have Not Received Prior Systemic Anti-cancer Treatment for Advanced Disease; ClinicalTrials.gov identifier: NCT04478266),67 which compared amcenestrant + palbociclib versus letrozole + palbociclib as first line in HR+/HER2- aBC; the trial did not meet its primary endpoint of improving PFS.68 Similarly, AMEERA-3 (An Open Label Randomized Phase 2 Trial of Amcenestrant [SAR439859], Versus Endocrine Monotherapy as Per Physician’s Choice in Patients With Estrogen Receptor-positive, HER2-Negative Locally Advanced or Metastatic Breast Cancer With Prior Exposure to Hormonal Therapies; ClinicalTrials.gov identifier: NCT04059484) did not meet its primary endpoint of improving PFS among endocrine-pretreated patients; therefore, the development of amcenestrant has been ceased (Table 1).48
Imlunestrant is another oral SERD with pure antagonistic activity, which has shown activity in preclinical studies, including in ESR1-mutant models.69 The EMBER-1 phase Ia/b trial (A Study of LY3484356 in Participants with Advanced or Metastatic Breast Cancer or Endometrial Cancer [EMBER-1]; ClinicalTrials.gov identifier: NCT04188548)70 looked at imlunestrant monotherapy in 114 patients with HR+/HER2- aBC who received three lines or less prior therapies for aBC. In evaluable patients, ORR was 8.0% (6/75) and CBR was 40.4% (42/104). Clinical benefit was observed regardless of baseline ESR1 mutation status. At the recommended phase II dose of 400 mg, the most common all-grade AEs were nausea (33.3%), fatigue (27.5%) and diarrhoea (23.2%).70 Another arm of the EMBER-1 trial is evaluating imlunestrant in combination with abemaciclib and AI in patients with HR+/HER2 aBC who have not yet received CDK4/6i.71 Table 2 shows on-going trials assessing the role of SERDs and other novel ETs as monotherapy or in combination with other agents.
Table 2: Selected ongoing trials for novel endocrine therapies in advanced hormone receptor-positive/human epidermal growth factor-negative breast cancer
| Class | Agent | ClinicalTrial.gov Identifier | Phase | Sample size | Study description | Primary endpoint |
| Oral SERD | Elacestrant | NCT05596409† (ELCIN) | II | Estimated N=80 | Elacestrant in HR+/HER2- aBC with 1-2 prior hormonal therapies but no prior CDK4/6i | PFS |
|
|
| NCT04791384† | Ib/II | Estimated N=44 | Abemaciclib + Elacestrant in HR+/HER2- aBC with brain metastases | AEs, overall intracranial response rate, CBR |
|
|
| NCT05386108† (ELECTRA) | Ib/II | Estimated N=106 | Abemaciclib + Elacestrant in HR+/HER2- aBC with or without brain metastases | R2PD, ORR |
|
|
| NCT05963997† (SUMIT-ELA) | Ib/II | Estimated N=48 | Samuraciclib + Elacestrant in HR+/HER2- aBC | RP2D, PFS |
|
|
| NCT06062498‡ | II | Estimated N=174 | Elacestrant vs. Elacestrant + CDK4/6i in HR+/HER2- aBC with prior CDK4/6i | PFS |
|
| Giredestrant | NCT04546009* (persevERA) | III | N=992 | Giredestrant + palbociclib vs. letrozole + palbociclib in HR+/HER2- aBC | PFS |
|
|
| NCT05306340† (evERA) | III | Estimated N=320 | Giredestrant + everolimus vs. exemestane + everolimus in ER+/HER2- aBC previously on CDK4/6i+ET | PFS in the ESR1m subpopulation and ITT population |
|
|
| NCT06065748‡ (pionERA) | III | Estimated N=1050 | Giredestrant vs. fulvestrant in combination with investigator’s choice CDK4/6i in HR+/HER2- aBC resistant to adjuvant ET | PFS in ESR1m subgroup and PFS in full analysis set population |
|
|
| NCT04802759† (MORPHEUS) | Ib/II | Estimated N=510 | Giredestrant vs. giredestrant + palbociclib or ribociclib in patients with disease progression on 1-2 lines of ET | Safety, ORR |
|
| Imlunestrant | NCT04975308† (EMBER-3) | III | Estimated N=860 | Imlunestrant ±abemaciclib vs. fulvestrant or exemestane in HR+/HER2- aBC on prior ET | PFS in ESR1m population and ITT population |
|
| Camizestrant | NCT04711252† (SERENA-4) | III | Estimated N=1342 | Camizestrant + Palbociclib vs. Anastrozole + Palbociclib in untreated ER+/HER2- aBC | PFS |
|
|
| NCT04964934† (SERENA-6) | III | Estimated N=300 | Camizestrant + CDK4/6i vs. AI + CDK4/6i in HR+/HER2- ESR1 mutated aBC | PFS |
| SERCA | H3B-6545 | NCT04288089* | Ib | Estimated N=36 | H3B-6545 + Palbociclib in HR+/HER2- aBC | MTD, RP2D |
| CERAN | OP-1250 | NCT05266105† | Ib | Estimated N=30 | OP-1250 + palbociclib in HR+/HER2- aBC | Safety, PK, plasma levels |
|
|
| NCT05508906† | Ib | Estimated N=60 | OP-1250 + ribociclib vs. OP-1250 + alpelisib in HR+/HER2- aBC with prior ET | Tolerability, safety, PK |
|
|
| NCT06016738‡ (OPERA-01) | III | Estimated N=510 | OP-1250 vs. SoC ET in HR+/HER2- aBC progression on ET + CDK4/6i | PFS in ESR1m and non-mutated patients |
| PROTAC | ARV-471 | NCT04072952† | I/II | Estimated N=215 | ARV-471 ± palbociclib in HR+/HER2- aBC with prior ET and chemotherapy | Safety, tolerability, anti-tumor activity |
|
|
| NCT05548127† NCT0557355† (TACTIVE-U) | Ib/2 | Estimated N=35 | ARV-471 + abemaciclib and ribociclib in HR+/HER2- aBC | Phase 1b: DLTs Phase 2: ORR |
|
|
| NCT05501769† (TACTIVE-E) | Ib | Estimated N=32 | ARV-471 + everolimus in HR+/HER2- aBC with prior CDK4/6i and ET | Safety, tolerability |
|
|
| NCT05654623† (VERITAC-2) | III | Estimated N=560 | ARV-471 vs fulvestrant in HR+/HER2- aBC after prior CDK4/6i and ET | PFS |
|
|
| NCT05909397† (VERITAC-3) | III | Estimated N=1180 | ARV-471 + palbociclib vs letrozole + palbociclib in untreated HR+/HER2- aBC | PFS |
| Third generation SERM | Lasofoxifene | NCT05696626‡ (ELAINEIII) | III | Estimated N=400 | Lasofoxifene + abemaciclib vs. fulvestrant + abemaciclib in HR+/HER2- ESR1m aBC with prior CDK4/6i-based treatment | PFS |
| SARM | Enobosarm | NCT04869943* (ARTEST) | III | Estimated N=210 | Enobosarm monotherapy vs exemestane in AR+/ER+/HER2- mBC with AR Staining, previously treated with AI, SERD & CDK4/6i | PFS |
|
|
| NCT05065411* (ENABLER-2) | III | Estimated N=186 | Enobosam + abemaciclib vs. fulvestrant in AR+/ER+/HER2- mBC, progressed on estrogen blocking agent + palbociclib | Safety, PFS |
|
| EP0062 | NCT05573126† | I/II | Estimated N=128 | EP0062 in ER+/AR+/HER2- relapsed aBC | DLTs, MTD, and AEs |
*Recruiting. †Not yet recruiting.‡ Complete. §Active, not recruiting.
Full names of clinical trials: ELCIN=Elacestrant in Women and Men With CDK4/6 Inhibitor-Naive Estrogen Receptor Positive, HER-2 Negative Metastatic Breast Cancer: An Open-Label Multicenter Phase 2 Study; ELECTRA=An Open-label Multicenter Phase 1b-2 Study of Elacestrant in Combination With Abemaciclib in Women and Men With Brain Metastasis From Estrogen Receptor Positive, HER-2 Negative Breast Cancer; SUMIT-ELA=A Phase 1b/2 Open-label Study of Samuraciclib in Combination With Elacestrant in Participants With Metastatic or Locally Advanced Hormone Receptor-positive and Human Epidermal Growth Factor Receptor 2-negative Breast Cancer; persevERA=A Phase III Randomized, Double-Blind, Placebo-Controlled, Multicenter Study Evaluating the Efficacy and Safety of GDC-9545 Combined With Palbociclib Compared With Letrozole Combined With Palbociclib in Patients With Estrogen Receptor-Positive, HER2-Negative Locally Advanced or Metastatic Breast Cancer; evERA=A Phase III, Randomized, Open-Label, Multicenter Study Evaluating the Efficacy and Safety of Giredestrant Plus Everolimus Compared With The Physician’s Choice of Endocrine Therapy Plus Everolimus in Patients With Estrogen Receptor-Positive, HER2-Negative, Locally Advanced or Metastatic Breast Cancer; pionERA=A Phase III Randomized, Open-Label Study Evaluating Efficacy and Safety of Giredestrant Compared With Fulvestrant, Both Combined With a CDK4/6 Inhibitor, in Patients With Estrogen Receptor-Positive, HER2-Negative Advanced Breast Cancer With Resistance to Prior Adjuvant Endocrine Therapy; MORPHEUS=A Phase Ib/II, Open-Label, Multicenter, Randomized Umbrella Study Evaluating the Efficacy and Safety of Multiple Treatment Combinations in Patients With Breast Cancer; EMBER-3=A Phase 3, Randomized, Open-Label Study of Imlunestrant, Investigator’s Choice of Endocrine Therapy, and Imlunestrant Plus Abemaciclib in Patients With Estrogen Receptor Positive, HER2 Negative Locally Advanced or Metastatic Breast Cancer Previously Treated With Endocrine Therapy; SERENA-4=A Randomised, Multicentre, Double-Blind, Phase III Study of AZD9833 (an Oral SERD) Plus Palbociclib Versus Anastrozole Plus Palbociclib for the Treatment of Patients With Estrogen Receptor-Positive, HER2-Negative Advanced Breast Cancer Who Have Not Received Any Systemic Treatment for Advanced Disease; SERENA-6=A Phase III, Double-blind, Randomised Study to Assess Switching to AZD9833 (a Next Generation, Oral SERD) + CDK4/6 Inhibitor vs Continuing Aromatase Inhibitor (Letrozole or Anastrozole) + CDK4/6 Inhibitor in HR+/HER2-MBC Patients With Detectable ESR1Mutation Without Disease Progression During 1L Treatment With Aromatase Inhibitor+ CDK4/6 Inhibitor- A ctDNA Guided Early Switch Study; OPERA-01=A Phase 3 Randomized, Open-Label Study of OP-1250 Monotherapy vs Standard of Care for the Treatment of ER+, HER2- Advanced or Metastatic Breast Cancer Following Endocrine and CDK 4/6 Inhibitor Therapy; TACTIVE-U=An Interventional Safety and Efficacy Phase 1b/2, Open-label Umbrella Study to Investigate Tolerability, pk, and Antitumor Activity of Vepdegestrant (ARV-471/PF-07850327), an Oral Proteolysis Targeting Chimera, in Combination With Other Anticancer Treatments in Participants Aged 18 Years and Over With ER+ Advanced or Metastatic Breast Cancer, Sub-study A,B,C; TACTIVE-E=A Phase 1b Trial of ARV-471 in Combination With Everolimus in Patients With ER+, HER2- Advanced or Metastatic Breast Cancer; VERITAC-2=A Phase 3, Randomized, Open-Label, Multicenter Trial of Arv-471 (Pf-07850327) Vs Fulvestrant in Participants with Estrogen Receptor-Positive, Her2-Negative Advanced Breast Cancer Whose Disease Progressed After Prior Endocrine Based Treatment for Advanced Disease; VERITAC-3=A Phase 3, Randomized, Open-Label, Multicenter Study of Arv-471(pf-07850327) Plus Palbociclib Versus Letrozole Plus Palbociclib for the Treatment of Participants with Estrogen Receptor-Positive, Her2-Negative Breast Cancer Who Have Not Received Any Prior Systemic Anti-Cancer Treatment for Advanced Disease; ELAINE III=An Open Label, Randomized, Multicenter Study Comparing the Efficacy and Safety of the Combination of Lasofoxifene and Abemaciclib to the Combination of Fulvestrant and Abemaciclib for the Treatment of Pre- and Postmenopausal Women and Men With Locally Advanced or Metastatic ER+/HER2- Breast Cancer With an ESR1 Mutation; ARTEST=Randomized Crossover Ph3 to Evaluate Efficacy/Safety of Enobosarm Monotherapy vs Active Control for Treatment of AR+/ER+/HER2- MBC With AR Staining Previously Treated w/Nonsteroidal Aromatase Inhibitor, SERD & CDK 4/6 Inhibitor; ENABLER=P3 Efficacy Evaluation of Enobosarm in Combo With Abemaciclib Compared to Estrogen Blocking Agent for 2nd Line Treatment of ER+HER2- MBC in Patients Who Have Shown Previous Disease Progression on an Estrogen Blocking Agent Plus Palbociclib.
Source for table and footnotes: ClinicalTrials.gov.72
aBC = advanced breast cancer; AE = adverse event; AI = aromatase inhibitor; AR = androgen receptor; CBR = clinical benefit rate; CDK4/6i = cyclin-dependent kinase 4/6 inhibitor; CERAN = complete oestrogen receptor antagonist; DLT = dose-limiting toxicity; ESR1m = oestrogen receptor 1 mutant; ET = endocrine therapy; HER2- = human epidermal growth factor negative; HR+ = hormone receptor positive; ITT = intention to treat; MTD = maximum tolerated dose; ORR = overall response rate; PFS = progression-free survival; PK = pharmacokinetics; PROTACs = proteolysis-targeting chimeras; R2PD = recommended phase II dose; SARM = selective androgen receptor modulator; SERCA = selective oestrogen receptor covalent antagonists; SERD = selective oestrogen receptor degrader; SERM = selective oestrogen receptor modulators; SoC = standard of care.
Oestrogen receptor antagonists (selective oestrogen receptor covalent antagonist and complete oestrogen receptor antagonist)
Similar to SERDs, selective oestrogen receptor covalent antagonist (SERCA) and complete oestrogen receptor antagonist (CERAN) are drugs that target the ER with the goal of inactivation and decreasing transcriptional activity.
SERCAs are a covalent class of ER antagonist that promotes an antagonistic conformation of both WT and ESR1-mutant ER by targeting a unique cysteine residue (Cys530) that is not found in other steroid hormone receptors (Figure 2).73 H3B-6545 is a SERCA that has shown preclinical and early clinical activity.74 A phase I/II study (Trial of H3B-6545, in Women With Locally Advanced or Metastatic Estrogen Receptor-positive, HER2 Negative Breast Cancer; ClinicalTrials.gov identifier: NCT03250676) evaluated H3B-6545 as monotherapy in 94 HR+/HER2- heavily pretreated patients with aBC (a median of three lines of prior therapy).75 In the evaluable population (N=72), ORR was 16.7% and CBR was 40.3%; mPFS in the overall population (n=94) was 5.1 months. However, in the ESR1 clonal Y537S subgroup (n=10), there was a better ORR of 30.0%, CBR of 60.0% and mPFS of 7.3 months. The tumours harbouring the ESR1 Y537S mutation have constitutionally active ER. As H3B-6545 antagonizes ER activity, this could explain the better activity in patients with ESR1 mutations. Notable AEs include grade 1 (34%) to 2 (5%) sinus bradycardia and grade 2 (2%) to 3 (3%) QTc (heart rate corrected QT interval) prolongation.75,76
CERANs such as OP-1250 fully inhibit the activity of both WT and mutant forms of ER by blocking transcriptional activation domain-activating factor 1 (AF1) and 2 (AF2). In contrast, SERMs block AF2 and incompletely antagonize AF1, which can be turned on via signalling pathways and has been shown to play a role in the development of endocrine resistance.77 In addition, OP-1250 also acts as a SERD by inducing ER degradation.77 In preclinical studies, OP-1250 in combination with CDK4/6i demonstrated synergistic activity in models of WT and ESR1-mutated ER, and in patients with brain metastases.78 A phase Ib/II trial of OP-1250 with palbociclib (A Phase 1 Study of OP-1250 in Combination With Palbociclib in HR+/ HER2- Breast Cancer Patients; ClinicalTrials.gov identifier: NCT05266105)79 displayed tolerability and induced tumour responses and disease stabilization in patients with HR+/HER2- aBC who received up to two lines of prior treatment. Out of 20 enrolled patients, 5 achieved a partial response (PR) and 10 had stable disease. The most common AEs included neutropenia, thrombocytopenia and gastrointestinal side effects (nausea, vomiting and diarrhoea). All were grade 1 to 2, except neutropenia, and grade 3 neutropenia occurred in 55% of patients.79
Proteolysis-targeting chimeras
Proteolysis-targeting chimeras (PROTACs) are molecules that contain both a domain that binds a target protein and a domain that binds an E3-ubiquitin ligase. The proximity of these two elements encourages ubiquitination and proteasomal degradation of the target protein.80
ARV-471 (vepdegestrant) is a selective, orally administered PROTAC that targets both WT and mutant ER. The phase II portion of the VERITAC trial (A Phase 1/2 Trial of ARV-471 Alone and in Combination With Palbociclib [IBRANCE®] in Patients With ER+/HER2- Locally Advanced or Metastatic Breast Cancer [mBC]; ClinicalTrials.gov identifier: NCT04072952)81 enrolled 71 patients with HR+/HER2- aBC who received one or more lines of prior ET for 6 months or more, one or more CDK4/6i and one or less chemotherapy regimen. CBR was 37.1% in 35 evaluable patients treated with 200 mg daily and 38.9% in 36 evaluable patients treated with 500 mg daily. CBR was better in evaluable patients with mutant ESR1. In the 200 mg ESR1-mutated cohort (n=19), CBR was 47.4, and 54.5% in the 500 mg ESR1-mutated cohort (n=22). ARV-471 was well tolerated at both doses (200 and 500 mg), with most AE grade 1 to 2 (most common were fatigue and nausea).81 Overall, ARV-471 monotherapy showed evidence of clinical activity based on CBR, which was further enhanced in the subgroup with ESR1 mutations.
Next-generation selective oestrogen receptor modulator
Lasofoxifene, a third-generation SERM, has a similar mechanism to tamoxifen but better oral bioavailability.82 In preclinical models with ESR1 mutations, lasofoxifene was shown to have superior efficacy over fulvestrant as a monotherapy or combined with CDK4/6i.77,82 The phase II ELAINE I trial (An Open-Label, Randomized, Multicenter Study Evaluating the Activity of Lasofoxifene Relative to Fulvestrant for the Treatment of Pre- and Postmenopausal Women With Locally Advanced or Metastatic ER+/HER2- Breast Cancer With an ESR1 Mutation; ClinicalTrials.gov identifier: NCT03781063) is summarized in Table 1.58 The phase II ELAINE II trial (An Open-label, Multicenter Study Evaluating the Safety of Lasofoxifene in Combination With Abemaciclib for the Treatment of Pre- and Postmenopausal Women With Locally Advanced or Metastatic ER+/HER2- Breast Cancer and Have an ESR1 Mutation; ClinicalTrials.gov identifier: NCT04432454)83 assessed lasofoxifene with abemaciclib in 29 patients with HR+/HER2- aBC with acquired ESR1 mutation who received a median of two prior lines of therapy (28/29 had prior CDK4/6i). The mPFS of this study was notable at 56.0 weeks. The most common AEs were diarrhoea, nausea and leucopenia, most likely attributable to abemaciclib.83 Overall, lasofoxifene in combination with CDK4/6i seems to have greater clinical benefit than when used alone.
Targeting the androgen receptor
The androgen receptor (AR) is another target of growing interest in aBC. Preclinical models have shown that AR activation has anti-tumour activity in ER+/AR+ BCs. AR activation leads to the alteration of the genomic distribution of ER and essential co-activators, resulting in the repression of ER-regulated cell cycle genes and upregulation of AR target genes, known as tumour suppressors.84 Enobosarm is a selective androgen receptor modulator (SARM). In a phase II clinical trial (A Phase 2 Open Label, Multi-Center, Multinational, Randomized, Parallel Design Study Investigating The Efficacy and Safety Of GTx-024 On Metastatic or Locally Advanced ER+/AR+ Breast Cancer [BC] in Postmenopausal Women; ClinicalTrials.gov identifier: NCT02463032)85 of 136 heavily pretreated patients with AR+/ER + aBC receiving enobosarm, patients with AR staining of >40% benefitted more. The best objective tumour response in patients with >40% AR was 48%, and <40% AR was 0% (p<0.00001). The median radiographic PFS was 5.47 months with >40% AR and 2.72 months for <40% AR.85 Enobosarm and other SARMs such as EP0062 are currently under investigation (Table 2).85,86
Targeting the phosphoinositide 3-kinase/AKT/mammalian target of rapamycin pathway
The PI3K/AKT pathway is important for cell growth and survival.25 Enhanced activation of this pathway represents an oncogenic driver and can determine resistance to ET in patients with HR+ BC, and several agents targeting this pathway have been approved.87
Phosphoinositide 3-kinase inhibitors
Class I PI3K has four isoforms based on catalytic domain: α (p110α/PI3Kα), β (p110β/PI3Kβ), δ (p110δ/PI3Kδ) and γ (p110γ/PI3Kγ).88 Alpelisib is an oral α-specific PI3K inhibitor that selectively inhibits p110α, thereby inhibiting the activation of the PI3K/AKT pathway.89 It was approved by the FDA in 2019 for patients with HR+/HER2-, PIK3CA-mutated aBC based on the results from the phase III SOLAR-1 trial (Study Assessing the Efficacy and Safety of Alpelisib Plus Fulvestrant in Men and Postmenopausal Women With Advanced Breast Cancer Which Progressed on or After Aromatase Inhibitor Treatment; ClinicalTrials.gov identifier: NCT02437318).8,90 The alpelisib + fulvestrant group had an mPFS of 11 versus 5.7 months in the fulvestrant group (HR 0.65; 95% CI 0.50–0.85; p<0.001).8 More severe (grade 3 to 4) AEs occurring in the alpelisib + fulvestrant group include hyperglycaemia (36.6 versus 0.7% in the control arm), rash (9.9 versus 0.3%) and diarrhoea (6.7 versus 0.3%). However, as CDK4/6i became the SoC after SOLAR-1 finished recruitment, the subsequent phase II BYLieve trial (Study Assessing the Efficacy and Safety of Alpelisib Plus Fulvestrant or Letrozole, Based on Prior Endocrine Therapy, in Patients With PIK3CA Mutant, HR+, HER2- Advanced Breast Cancer Who Have Progressed on or After Prior Treatments; ClinicalTrials.gov identifier: NCT03056755) was performed.49 Cohort A and B patients were treated with alpelisib + fulvestrant and alpelisib + letrozole, respectively, and both cohorts had CDK4/6i + ET (AI in cohort A and fulvestrant in cohort B) as prior treatment. Cohort C enrolled patients previously treated with chemotherapy or ET. Based on the long-term follow-up, mPFS in cohorts A, B and C was 8.0, 5.6 and 5.6 months, respectively.49 In Table 3, we summarize on-going studies using novel targeted agents for HR+/HER2- aBC.
Table 3: Selected ongoing trials for non-endocrine targets in advanced hormone receptor-positive/human epidermal growth factor 2-negative breast cancer
| Class | Agent | Clinical trial identifier | Phase | Sample size | Study description | Primary endpoint |
| PI3K inhibitor | Alpelisib | NCT05038735† (EPIK-B5) | III | Estimated N=234 | Alpelisib + fulvestrant versus placebo + fulvestrant for men and postmenopausal women with HR+/HER2- PIK3CAm aBC, progressed on or after AI and CDK4/6i | PFS |
| NCT05625087‡ (SAFIR 03) | II | Estimated N=162 | Alpelisib + fulvestrant versus ribociclib + fulvestrant in HR+/HER2- PIK3CAm aBC | PFS | ||
| NCT02379247§ | I/II | N=43 | Alpelisib + nab-paclitaxel in HER2- aBC | RP2D and ORR | ||
| NCT04762979† | II | Estimated N=44 | Alpelisib + fulvestrant or AI in HR+/HER2- PIK3CAm aBC, progressed on ET | PFS | ||
| NCT02058381§ (B-YOND) | Ib | N=40 | Tamoxifen + goserelin acetate with alpelisib or buparlisib in HR+/HER2- aBC | MTD and RP2D | ||
| NCT05392608† (SEQUEL-breast) | II | Estimated N=130 | Fulvestrant + alpelisib after progression on fulvestrant (±prior CDK4/6i) in HR+/HER2- PIK3CAm aBC | PFS | ||
| Inavolisib | NCT05646862† (INAVO121) | III | Estimated N=400 | Inavolisib + fulvestrant versus alpelisib + fulvestrant HR+/HER2-, PIK3CAm aBC/mBC, progressed during or after CDK4/6i therapy | PFS | |
| PI3K and mTOR inhibitor | Gedatolisib | NCT02684032§ | Ib | N=141 | Gedatolisib + palbociclib and AI in HR+/HER2- aBC | DLTs and ORR |
| NCT05501886† (VIKTORIA-1) | III | Estimated N=701 | Gedatolisib + fulvestrant ± palbociclib in HR+/HER2- aBC progressed on CDK4/6i and AI | PFS in PIK3CA WT and mutant BC | ||
| PIK3CA-mutant inhibitor | LOXO-783 | NCT05307705† (PIKASSO-01) | Ib | Estimated N=400 | LOXO-783 alone and in combination with fulvestrant, imlunestrant, abemaciclib, AI or paclitaxel in aBC with PIK3CA H1047R mutation | MTD, RP2D and DLTs |
| STX-478 | NCT05768139† | I/II | Estimated N=220 | STX-478 alone and in combination with fulvestrant in aBC and other solid tumours | Safety, tolerability, PK and preliminary anti-tumour activity | |
| RLY-2608 | NCT05216432† | Ib | Estimated N=235 | RLY-2608 alone and in combination with fulvestrant in HR+/HER2- aBC | MTD, RP2D and AEs | |
| AKT inhibitor | Capivasertib | NCT04862663† (CAPItello-292) | Ib/III | Estimated N=850 | Capivasertib + CDK4/6i + fulvestrant versus CDK4/6i + fulvestrant in HR+/HER2- aBC without prior ET or CDK4/6i | Phase Ib: DLTs and AEs Phase III: PFS |
| NCT01625286§ (BEECH) | Ib/III | N=148 | Capivasertib + paclitaxel versus paclitaxel + placebo in ER+ aBC (subgroup with PIK3CAm) | DLTs and PFS | ||
| NCT05720260† | II | Estimated N=56 | Capivasertib + goserelin + fulvestrant with/without durvalumab versus goserelin + fulvestrant + durvalumab versus goserelin + fulvestrant in ER+ aBC failed on two lines of ET | PFS | ||
| Ipatasertib | NCT04920708† (FAIM) | II | Estimated N=324 | Ipatasertib + fulvestrant + palbociclib versus palbociclib + fulvestrant in HR+/HER2- aBC without ctDNA suppression | PFS | |
| NCT04650581† (FINER) | III | Estimated N=250 | Ipatasertib + fulvestrant versus placebo + fulvestrant in ER+/HER2- aBC, progressed on CDK4/6i and AI | PFS | ||
| NCT04802759† | Ib/II | Estimated N=510 | Umbrella study, cohort 1: ipatasertib treatment combinations in HR+/HER2- aBC progressed on CDK4/6i | ORR and AEs | ||
| NCT03424005† (MORPHEUS-panBC) | Ib/II | Estimated N=242 | Ipatasertib combinations in aBC (cohort 3 HR+ and HER2-negative disease with PIK3CA mutation) | ORR and AEs | ||
| CDK4 inhibitor | PF-07220060 | NCT05262400† | Ib/II | Estimated N=240 | PF-07220060 + PF-07104091 + ET in HR+/HER2- BC and other solid tumours | DLTs and AEs |
| CDK2 inhibitor | PF-07104091 | NCT04553133† | I/IIa | Estimated N=320 | PF-07104091 monotherapy in HR+/HER2- aBC on two or more lines of treatment including ET and CDK4/6i | MTD and/or RP2D, DLTs and AEs |
| ARTS-021 | NCT05867251† | I/II | Estimated N=192 | ARTS-021 alone and in combination with ET + CDK4/6i in HR+/HER2- aBC unresponsive to standard therapy | DLTs, AEs, RP2D, ORR, PFS, OS and TPP | |
| BLU-222 | NCT05252416† (VELA) | I/II | Estimated N=366 | BLU-222 alone and in combination with carboplatin, ribociclib or fulvestrant in HR+/HER2- BC, progressed on CDK4/6i and other advanced solid tumours | MTD, RP2D, AEs and ORR | |
| CDK7 inhibitor | Samuraciclib | NCT05963984† (SUMIT-BC) | II | Estimated N=60 | Samuraciclib + fulvestrant versus fulvestrant alone in HR+/HER2- aBC | CBR |
| NCT05963997† (SUMIT-ELA) | Ib/II | Estimated N=48 | Samuraciclib + elacestrant in HR+/HER2- aBC | Phase Ib: RP2D and AEs Phase II: PFS |
*Active, not recruiting. †Recruiting. ‡Not yet recruiting. §Complete.
Full names of clinical trials: EPIK-B5=A Phase III, Randomized, Double-blind, Placebo-controlled Study of Alpelisib in Combination With Fulvestrant for Men and Postmenopausal Women With HR-positive, HER2-negative Advanced Breast Cancer With a PIK3CA Mutation, Who Progressed on or After Aromatase Inhibitor and a CDK4/6 Inhibitor; SAFIR-03=A ctDNA Screening Program in Patients With HR+, HER2- Metastatic Breast Cancer for Detection of High-risk Relapse Patients on Any CDK4/6 Inhibitor and a Randomised Phase II Study Comparing Alpelisib Combined With Fulvestrant to Ribociclib Combined With Fulvestrant, in Patients With Persistent Targetable PIK3CA Mutations; B-YOND=A Phase Ib Dose De-escalation Study of the Combination of Tamoxifen Plus Goserelin Acetate With Alpelisib (BYL719) or Buparlisib (BKM120) in Premenopausal Patients With Hormone Receptor-positive/HER2-negative Locally Advanced or Metastatic Breast Cancer; SEQUEL-breast=SEQUence of Endocrine Therapy in Advanced Luminal Breast Cancer (SEQUEL-Breast): A Phase 2 Study on Fulvestrant Beyond Progression in Combination With Alpelisib for PIK3CA-mutated, Hormone-receptor Positive HER2 Negative Advanced Breast Cancer; INAVO-121=A Phase III, Multicenter, Randomized, Open-Label Study Evaluating the Efficacy and Safety of Inavolisib Plus Fulvestrant Versus Alpelisib Plus Fulvestrant in Patients With Hormone Receptor-Positive, HER2-Negative, PIK3CA Mutated, Locally Advanced or Metastatic Breast Cancer Who Progressed During or After CDK4/6 Inhibitor and Endocrine Combination Therapy; VIKTORIA-1=Phase 3, Open-Label, Randomized, Study Comparing Gedatolisib Combined With Fulvestrant & With or Without Palbociclib to Standard-of-Care Therapies in Patients With HR-Positive, HER2-Negative Advanced Breast Cancer Previously Treated With a CDK4/6 Inhibitor in Combination w/Non-Steroidal Aromatase Inhibitor Therapy; PIKASSO-01=A Study of LOXO-783 Administered as Monotherapy and in Combination With Anticancer Therapies for Patients With Advanced Breast Cancer and Other Solid Tumors With a PIK3CA H1047R Mutation; CAPItello-292=A Phase Ib/III, Open-label, Randomised Study of Capivasertib Plus CDK4/6 Inhibitors and Fulvestrant Versus CDK4/6 Inhibitors and Fulvestrant in Hormone Receptor-Positive and Human Epidermal Growth Factor Receptor 2-Negative Locally Advanced, Unresectable or Metastatic Breast Cancer; BEECH=A Phase I/II Study of AZD5363 Combined With Paclitaxel in Patients With Advanced or Metastatic Breast Cancer. Comprising a Safety Run-In and a Placebo-controlled Randomised Expansion in ER+ve Patients Stratified by PIK3CA Mutation Status; FAIM=Randomised Phase II Study of Induction Fulvestrant and CDK4/6 Inhibition With the Addition of Ipatasertib in Metastatic ER+/HER2- Breast Cancer Patients Without ctDNA Suppression; FINER=Double-Blind Placebo-Controlled Randomized Phase III Trial of Fulvestrant and Ipatasertib as Treatment for Advanced HER-2 Negative and Estrogen Receptor Positive (ER+) Breast Cancer Following Progression on First Line CDK 4/6 Inhibitor and Aromatase Inhibitor; MORPHEUS-panBC=A Phase Ib/II, Open-Label, Multicenter, Randomized Umbrella Study Evaluating The Efficacy And Safety Of Multiple Treatment Combinations In Patients With Metastatic Breast Cancer; VELA=A Phase 1/2 Study to Evaluate the Safety, Pharmacokinetics, and Efficacy of BLU-222 as a Single Agent and in Combination Therapy for Patients With Advanced Solid Tumors; SUMIT-BC=An Open-label, Interventional, Multicenter, Randomized, Phase 2 Study of Fulvestrant With or Without Samuraciclib in Participants With Metastatic or Locally Advanced Hormone Receptor (HR) Positive and Human Epidermal Growth Factor Receptor (HER)2-Negative Breast Cancer (BC); SUMIT-ELA=A Phase 1b/2 Open-label Study of Samuraciclib in Combination With Elacestrant in Participants With Metastatic or Locally Advanced Hormone Receptor-positive and Human Epidermal Growth Factor Receptor 2-negative Breast Cancer.
Source for table and footnotes: ClinicalTrials.gov.72
aBC = advanced breast cancer; AE = adverse event; AI = aromatase inhibitor; BC = breast cancer; CBR = clinical benefit rate; CDK4/6i = cyclin-dependent kinase 4/6 inhibitor; ctDNA = circulating tumour DNA; DLT = dose-limiting toxicity; ET = endocrine therapy; HER2- = human epidermal growth factor negative; HR+ = hormone receptor positive; MTD = maximum tolerated dose; mTOR = mammalian target of rapamycin; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; PI3K = phosphoinositide 3-kinase; PIK3CAm = phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha mutant; PK = pharmacokinetics; R2PD = recommended phase II dose; TTP = time to tumour progression; WT = wild type.
Inavolisib inhibits the PI3K/AKT pathway by binding to the adenosine triphosphate (ATP)-binding site of PI3Kα, blocking phosphorylation of PIP2/PIP3 and preventing downstream signalling. It also facilitates the degradation of mutant p110α, which may drive more potent and sustained PI3K inhibition.91 The phase III INAVO120 trial (A Phase III, Randomized, Double-Blind, Placebo-Controlled Study Evaluating the Efficacy and Safety of Inavolisib Plus Palbociclib and Fulvestrant Versus Placebo Plus Palbociclib and Fulvestrant in Patients With PIK3CA-Mutant, Hormone Receptor-Positive, Her2-Negative, Locally Advanced or Metastatic Breast Cancer; ClinicalTrials.gov identifier: NCT04191499)50 assessed inavolisib or placebo with palbociclib + fulvestrant in patients with PIK3CA-mutated, HR+ aBC which recurred during/within 12 months of completing adjuvant ET without prior treatment for aBC. The primary analysis was promising, showing an mPFS of 15 months in the inavolisib group versus 7.3 months in the placebo group (HR 0.43; 95% CI 0.32–0.59, p≤0.0001). All-grade AEs more common in the inavolisib group were consistent with known side effects of the class, including hyperglycaemia (58.6 versus 8.6% in the placebo group), diarrhoea (48.1 versus 16%) and rash (25.3 versus 17.3%).50
Gedatolisib is a dual PI3K/mTOR inhibitor. It is different from other agents targeting PI3K as it selectively targets all class I isoforms of PI3K and can potentially limit the development of drug resistance when compared with other isoform-specific PI3K inhibitors.92 The activation of the PI3K/AKT/mTOR pathway is also one way in which CDK4/6i resistance can develop; therefore, combination therapy with PI3K/mTOR inhibitor with a CDK4/6i could help restore sensitivity.92
PIK3 inhibitors have been associated with significant toxicity profiles. One of the most common on-target side effects is hyperglycaemia. Many cellular responses to insulin are mediated by the p110α catalytic subunit of PI3K and its downstream effectors.93 Inhibition of p110α blocks insulin signalling, leading to glycogen breakdown in the liver and decreased glucose uptake in peripheral tissue. This results in a transitory state of insulin resistance and hyperglycaemia. Insulin should be avoided in PI3K inhibitor-associated hyperglycaemia.94 Instead, metformin up to 2,000 mg daily can be used. If the condition persists, consultation with an endocrinologist to discuss the additional insulin sensitizer medications, such as pioglitazone, can be considered.95 PI3Kα is also involved in epithelial cell proliferation, maturation and apoptosis; therefore, rash and diarrhoea are also common AEs seen with PI3Kα inhibitors.96,97
The compounds under investigation that can potentially mitigate the toxicity related to the inhibition of WT PI3Kα (such as hyperglycaemia, skin rash and diarrhoea) include novel allosteric PIK3CA-mutant-specific inhibitors, such as LOXO-783. LOXO-783 is highly selective for PI3Kα H1047R and induced tumour regressions in HR+/HER2, PI3Kα H1047-mutant cancer models without inducing hyperglycaemia. LOXO-783 has shown additive effects when combined with fulvestrant or imlunestrant, and it appears to be effective in abemaciclib- and abemaciclib/fulvestrant-resistant models and have central nervous system (CNS) penetrance.98 RLY-2608 (A First-in-Human Study of Mutant-selective PI3Kα Inhibitor, RLY-2608, as a Single Agent in Advanced Solid Tumor Patients and in Combination With Fulvestrant in Patients With Advanced Breast Cancer; ClinicalTrials.gov identifier: NCT05216432) and STX-478 (First-in-Human Study of STX-478, a Mutant-Selective PI3Kα Inhibitor as Monotherapy and in Combination With Other Antineoplastic Agents in Participants With Advanced Solid Tumors; ClinicalTrials.gov identifier: NCT05768139) are other mutant-selective PI3Kα inhibitors under phase I/II investigation.99,100
AKT inhibitors
Capivasertib is a first-in-class AKT inhibitor approved by the FDA in November 2023 in combination with fulvestrant for patients with HR+/HER2- aBC who had received at least one prior line of ET and have somatic PIK3CA, AKT1 or PTEN alterations.101 CAPItello-291 (A Phase III Double-blind Randomised Study Assessing the Efficacy and Safety of Capivasertib + Fulvestrant Versus Placebo + Fulvestrant as Treatment for Locally Advanced [Inoperable] or Metastatic Hormone Receptor Positive, Human Epidermal Growth Factor Receptor 2 Negative [HR+/HER2-] Breast Cancer Following Recurrence or Progression On or After Treatment With an Aromatase Inhibitor; ClinicalTrials.gov identifier: NCT04305496) was a phase III trial that evaluated capivasertib or placebo with fulvestrant in 708 patients with HR+/HER2- aBC who progressed on AI.51 In the overall population, 69.1% received prior CDK4/6i as therapy for aBC and 289 patients (40.8%) had tumours with AKT pathway alterations. In the AKT pathway-altered group, mPFS was 7.3 versus 3.1 months in the capivasertib and control groups, respectively (HR 0.5; 95% CI 0.38–0.65; p≤0.001). This benefit did not translate into the AKT pathway non-altered group, with an mPFS of 5.3 months in the capivasertib group and 3.7 months in the control (HR 0.79; 95% CI 0.61–1.02). The most frequently reported grade ≥3 AEs were rash (12.1% in the capivasertib arm versus 0.3% in the control), diarrhoea (9.3 versus 0.3%) and hyperglycaemia (2.3 versus 0.3%).51 Ipatasertib is another AKT inhibitor under investigation, although it has not reached its primary endpoint of improving PFS in the HR+/HER2- aBC setting (IPATunity130 [A Double-Blind, Placebo-Controlled, Randomized Phase III Study of Ipatasertib in Combination With Paclitaxel as a Treatment for Patients With PIK3CA/AKT1/PTEN-Altered, Locally Advanced or Metastatic, Triple-Negative Breast Cancer or Hormone Receptor-Positive, HER2-Negative Breast Cancer]; ClinicalTrials.gov identifier: NCT03337724). Results for Cohort B are shown in Table 1.55
Novel cyclin-dependent kinase (cyclin-dependent kinase 2, cyclin-dependent kinase 4 and cyclin-dependent kinase 7) inhibitors
CDK4/6 inhibitors (ribociclib, palbociclib and abemaciclib) are the established first-line therapy in metastatic HR+ BC and have significantly improved outcomes for this disease.102 However, pharmacological attempts are ongoing to target additional components of the CDK family or to increase the selectivity of CDK4 inhibition to improve the activity and tolerability profiles of these agents.
Samuraciclib is an ATP-competitive inhibitor of CDK7. One arm of a phase I clinical trial (A Modular, Multipart, Multiarm, Open-label, Phase I/II Study to Evaluate the Safety and Tolerability of CT7001 Alone and in Combination With Anti-cancer Treatments in Patients With Advanced Malignancies; ClinicalTrials.gov identifier: NCT03363893) evaluated samuraciclib + fulvestrant in 31 patients with HR+/HER2- aBC who progressed on CDK4/6i therapy. The results in 25 evaluable patients showed was CBR 36% and mPFS was 3.7 months. Based on univariate analysis, mPFS was better in patients without a TP53 mutation (7.4 months). Ongoing preclinical work suggests that CDK7 inhibition can enhance the activity of TP53, which may explain the poorer response in the TP53-mutant cohort. The most common AEs present in ≥10% of patients were gastrointestinal side effects (diarrhoea, nausea and vomiting).103
CDK2 is also being investigated as a target, given CDK2/cyclin E is one method by which CDK4/6 resistance occurs.35 In preclinical trials for CDK2-selective inhibitor BLU-222, xenograft models of CDK4/6 resistance with cyclin E1 (CCNE1) overexpression showed resistance to ribociclib, but treatment with BLU-222 led to tumour regression.34 ARTS-021, another CDK2 inhibitor, has shown promising preclinical data in CCNE1-amplified patient-derived xenograft models.52
PF-07220060 is a CDK4-selective inhibitor that spares CDK6 and has shown promising activity in tumours that progressed on prior CDK4/6i. A phase I/II study (A Phase 1/2a Study Evaluating the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Anti-Tumor Activity of PF-07220060 as a Single Agent and as Part of Combination Therapy in Participants With Advanced Solid Tumors; ClinicalTrials.gov identifier: NCT04557449) is on-going in patients with advanced solid tumours.104 One arm of the study enrolled 26 patients with HR+/HER2- aBC who received a median 4.5 lines of prior therapy, including CDK4/6i. They received PF-07220060 (300 or 400 mg twice daily) in combination with letrozole or fulvestrant. Measurable responses were observed in six (28.6%) patients (one complete response (CR) and five PRs). CBR was seen in 11 out of 21 (52.4%) patients, and the mPFS was 24.7 weeks. The most common AEs were diarrhoea, neutropaenia and nausea with no grade >3 AEs.104
Discussion
ET is the backbone of treatment in aBC. However, most tumours develop endocrine resistance. There are several new agents under clinical development for the treatment of endocrine-resistant HR+/HER2- aBC that aim to overcome these mechanisms of resistance.
The introduction of these novel medications aims to give patients and providers more options, leading to improved cancer-related outcomes while maintaining a good quality of life. However, with the development of these new agents, an important question that remains is the optimal sequencing for endocrine and novel therapies. Currently, most patients are treated with CDK4/6i + ET, and at the time of progression, transition to other agents such as alpelisib, capivasertib or everolimus in combination with ET can be considered. Continuation of CDK4/6i while changing the ET backbone has been studied with mixed results, and it is not currently the SoC.105,106
The timing of somatic testing (liquid or tumour biopsy) has changed with the approval of alpelisib, elacestrant and capivasertib. Since ESR1 mutations are often acquired, testing after recurrence or progression on endocrine treatment is recommended when elacestrant is being considered.107 As seen in Table 1, studies assessing the role of oral SERDs have shown mixed results, and more studies are needed to determine the optimal application of SERDs and to find out the agents they can be combined with.
Many of these trials are testing ET in combination with other novel agents as well as monotherapy. While combined therapeutic strategies may help overcome treatment resistance and potentially re-sensitize tumours to ET, compounded AEs must also be considered, particularly given the target population are those living with metastatic disease and the preservation of quality of life should also be a goal.
Another challenge clinicians will face in an era with many developing therapies is that SoC may change from the time of trial recruitment to the trial end. This happened with the SOLAR-1 trial with alpelisib, where CDK4/6i became standard after the trial completed recruiting, and new trials had to be designed to verify the results. This fast-changing treatment landscape can make trials more difficult to design and results difficult to interpret.
In summary, there are multiple novel agents on the horizon for the treatment of HR+/HER2- aBC. Two agents were approved in 2023, such as elacestrant and capivasertib. More research is needed to determine optimal treatment combinations and sequencing, as well as to evaluate these agents as earlier lines of treatment for aBC or in the early HR+/HER2- BC setting. More work also needs to be done to determine the safety of these agents in combination with HER2-directed agents for the treatment of patients with HR+/HER2+ disease.